Nuriye Özlem Özcan Şimşek, Arzucan Özgür, Fikret Gürgen
{"title":"GNNMutation:一个基于异构图的癌症检测框架。","authors":"Nuriye Özlem Özcan Şimşek, Arzucan Özgür, Fikret Gürgen","doi":"10.1186/s12859-025-06133-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>When genes are translated into proteins, mutations in the gene sequence can lead to changes in protein structure and function as well as in the interactions between proteins. These changes can disrupt cell function and contribute to the development of tumors. In this study, we introduce a novel approach based on graph neural networks that jointly considers genetic mutations and protein interactions for cancer prediction. We use DNA mutations in whole exome sequencing data and construct a heterogeneous graph in which patients and proteins are represented as nodes and protein-protein interactions as edges. Furthermore, patient nodes are connected to protein nodes based on mutations in the patient's DNA. Each patient node is represented by a feature vector derived from the mutations in specific genes. The feature values are calculated using a weighting scheme inspired by information retrieval, where whole genomes are treated as documents and mutations as words within these documents. The weighting of each gene, determined by its mutations, reflects its contribution to disease development. The patient nodes are updated by both mutations and protein interactions within our noval heterogeneous graph structure. Since the effects of each mutation on disease development are different, we processed the input graph with attention-based graph neural networks.</p><p><strong>Results: </strong>We compiled a dataset from the UKBiobank consisting of patients with a cancer diagnosis as the case group and those without a cancer diagnosis as the control group. We evaluated our approach for the four most common cancer types, which are breast, prostate, lung and colon cancer, and showed that the proposed framework effectively discriminates between case and control groups.</p><p><strong>Conclusions: </strong>The results indicate that our proposed graph structure and node updating strategy improve cancer classification performance. Additionally, we extended our system with an explainer that identifies a list of causal genes which are effective in the model's cancer diagnosis predictions. Notably, some of these genes have already been studied in cancer research, demonstrating the system's ability to recognize causal genes for the selected cancer types and make predictions based on them.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"153"},"PeriodicalIF":3.3000,"publicationDate":"2025-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12139269/pdf/","citationCount":"0","resultStr":"{\"title\":\"GNNMutation: a heterogeneous graph-based framework for cancer detection.\",\"authors\":\"Nuriye Özlem Özcan Şimşek, Arzucan Özgür, Fikret Gürgen\",\"doi\":\"10.1186/s12859-025-06133-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>When genes are translated into proteins, mutations in the gene sequence can lead to changes in protein structure and function as well as in the interactions between proteins. These changes can disrupt cell function and contribute to the development of tumors. In this study, we introduce a novel approach based on graph neural networks that jointly considers genetic mutations and protein interactions for cancer prediction. We use DNA mutations in whole exome sequencing data and construct a heterogeneous graph in which patients and proteins are represented as nodes and protein-protein interactions as edges. Furthermore, patient nodes are connected to protein nodes based on mutations in the patient's DNA. Each patient node is represented by a feature vector derived from the mutations in specific genes. The feature values are calculated using a weighting scheme inspired by information retrieval, where whole genomes are treated as documents and mutations as words within these documents. The weighting of each gene, determined by its mutations, reflects its contribution to disease development. The patient nodes are updated by both mutations and protein interactions within our noval heterogeneous graph structure. Since the effects of each mutation on disease development are different, we processed the input graph with attention-based graph neural networks.</p><p><strong>Results: </strong>We compiled a dataset from the UKBiobank consisting of patients with a cancer diagnosis as the case group and those without a cancer diagnosis as the control group. We evaluated our approach for the four most common cancer types, which are breast, prostate, lung and colon cancer, and showed that the proposed framework effectively discriminates between case and control groups.</p><p><strong>Conclusions: </strong>The results indicate that our proposed graph structure and node updating strategy improve cancer classification performance. Additionally, we extended our system with an explainer that identifies a list of causal genes which are effective in the model's cancer diagnosis predictions. Notably, some of these genes have already been studied in cancer research, demonstrating the system's ability to recognize causal genes for the selected cancer types and make predictions based on them.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"153\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-06-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12139269/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06133-0\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06133-0","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
GNNMutation: a heterogeneous graph-based framework for cancer detection.
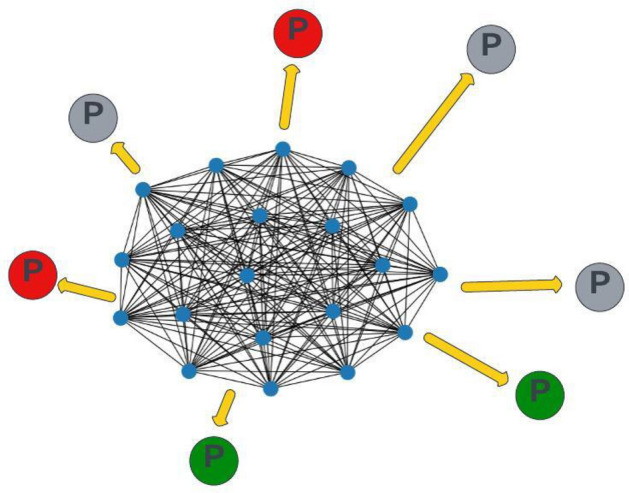
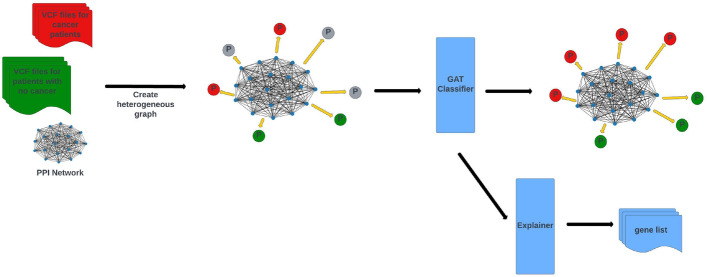
Background: When genes are translated into proteins, mutations in the gene sequence can lead to changes in protein structure and function as well as in the interactions between proteins. These changes can disrupt cell function and contribute to the development of tumors. In this study, we introduce a novel approach based on graph neural networks that jointly considers genetic mutations and protein interactions for cancer prediction. We use DNA mutations in whole exome sequencing data and construct a heterogeneous graph in which patients and proteins are represented as nodes and protein-protein interactions as edges. Furthermore, patient nodes are connected to protein nodes based on mutations in the patient's DNA. Each patient node is represented by a feature vector derived from the mutations in specific genes. The feature values are calculated using a weighting scheme inspired by information retrieval, where whole genomes are treated as documents and mutations as words within these documents. The weighting of each gene, determined by its mutations, reflects its contribution to disease development. The patient nodes are updated by both mutations and protein interactions within our noval heterogeneous graph structure. Since the effects of each mutation on disease development are different, we processed the input graph with attention-based graph neural networks.
Results: We compiled a dataset from the UKBiobank consisting of patients with a cancer diagnosis as the case group and those without a cancer diagnosis as the control group. We evaluated our approach for the four most common cancer types, which are breast, prostate, lung and colon cancer, and showed that the proposed framework effectively discriminates between case and control groups.
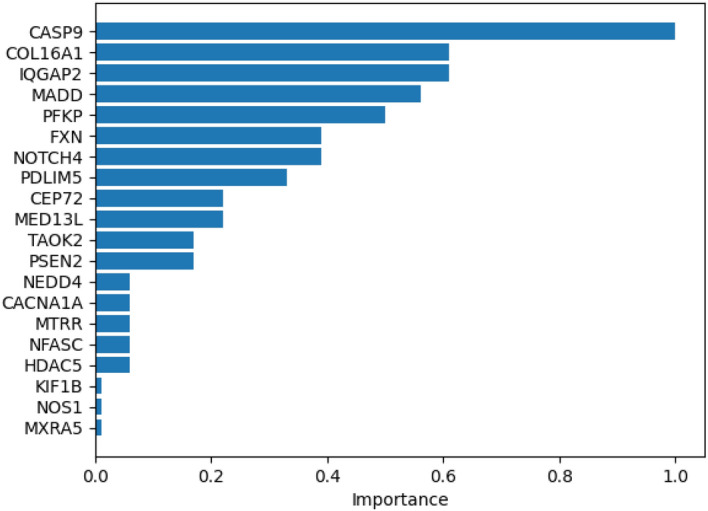
Conclusions: The results indicate that our proposed graph structure and node updating strategy improve cancer classification performance. Additionally, we extended our system with an explainer that identifies a list of causal genes which are effective in the model's cancer diagnosis predictions. Notably, some of these genes have already been studied in cancer research, demonstrating the system's ability to recognize causal genes for the selected cancer types and make predictions based on them.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


