Maryam Yousefian, Anna-Simone Frank, Marcus Weber, Susanna Röblitz
{"title":"基于区域分解的随机基因调控网络马尔可夫状态模型的高效构建。","authors":"Maryam Yousefian, Anna-Simone Frank, Marcus Weber, Susanna Röblitz","doi":"10.1186/s12859-025-06174-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The dynamics of many gene regulatory networks (GRNs) is characterized by the occurrence of metastable phenotypes and stochastic phenotype switches. The chemical master equation (CME) is the most accurate description to model such stochastic dynamics, whereby the long-time dynamics of the system is encoded in the spectral properties of the CME operator. Markov State Models (MSMs) provide a general framework for analyzing and visualizing stochastic multistability and state transitions based on these spectral properties. Until now, however, this approach is either limited to low-dimensional systems or requires the use of high-performance computing facilities, thus limiting its usability.</p><p><strong>Results: </strong>We present a domain decomposition approach (DDA) that approximates the CME by a stochastic rate matrix on a discretized state space and projects the multistable dynamics to a lower dimensional MSM. To approximate the CME, we decompose the state space via a Voronoi tessellation and estimate transition probabilities by using adaptive sampling strategies. We apply the robust Perron cluster analysis (PCCA+) to construct the final MSM. Measures for uncertainty quantification are incorporated. As a proof of concept, we run the algorithm on a single PC and apply it to two GRN models, one for the genetic toggle switch and one describing macrophage polarization. By comparing the results with reference solutions, we demonstrate that our approach correctly identifies the number and location of metastable phenotypes with adequate accuracy and uncertainty bounds. We show that accuracy mainly depends on the total number of Voronoi cells, whereas uncertainty is determined by the number of sampling points.</p><p><strong>Conclusions: </strong>A DDA enables the efficient computation of MSMs with quantified uncertainty. Since the algorithm is trivially parallelizable, it can be applied to larger systems, which will inevitably lead to new insights into cell-regulatory dynamics.</p>","PeriodicalId":8958,"journal":{"name":"BMC Bioinformatics","volume":"26 1","pages":"147"},"PeriodicalIF":3.3000,"publicationDate":"2025-06-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12131593/pdf/","citationCount":"0","resultStr":"{\"title\":\"Efficient construction of Markov state models for stochastic gene regulatory networks by domain decomposition.\",\"authors\":\"Maryam Yousefian, Anna-Simone Frank, Marcus Weber, Susanna Röblitz\",\"doi\":\"10.1186/s12859-025-06174-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The dynamics of many gene regulatory networks (GRNs) is characterized by the occurrence of metastable phenotypes and stochastic phenotype switches. The chemical master equation (CME) is the most accurate description to model such stochastic dynamics, whereby the long-time dynamics of the system is encoded in the spectral properties of the CME operator. Markov State Models (MSMs) provide a general framework for analyzing and visualizing stochastic multistability and state transitions based on these spectral properties. Until now, however, this approach is either limited to low-dimensional systems or requires the use of high-performance computing facilities, thus limiting its usability.</p><p><strong>Results: </strong>We present a domain decomposition approach (DDA) that approximates the CME by a stochastic rate matrix on a discretized state space and projects the multistable dynamics to a lower dimensional MSM. To approximate the CME, we decompose the state space via a Voronoi tessellation and estimate transition probabilities by using adaptive sampling strategies. We apply the robust Perron cluster analysis (PCCA+) to construct the final MSM. Measures for uncertainty quantification are incorporated. As a proof of concept, we run the algorithm on a single PC and apply it to two GRN models, one for the genetic toggle switch and one describing macrophage polarization. By comparing the results with reference solutions, we demonstrate that our approach correctly identifies the number and location of metastable phenotypes with adequate accuracy and uncertainty bounds. We show that accuracy mainly depends on the total number of Voronoi cells, whereas uncertainty is determined by the number of sampling points.</p><p><strong>Conclusions: </strong>A DDA enables the efficient computation of MSMs with quantified uncertainty. Since the algorithm is trivially parallelizable, it can be applied to larger systems, which will inevitably lead to new insights into cell-regulatory dynamics.</p>\",\"PeriodicalId\":8958,\"journal\":{\"name\":\"BMC Bioinformatics\",\"volume\":\"26 1\",\"pages\":\"147\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-06-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12131593/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s12859-025-06174-5\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s12859-025-06174-5","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Efficient construction of Markov state models for stochastic gene regulatory networks by domain decomposition.
Background: The dynamics of many gene regulatory networks (GRNs) is characterized by the occurrence of metastable phenotypes and stochastic phenotype switches. The chemical master equation (CME) is the most accurate description to model such stochastic dynamics, whereby the long-time dynamics of the system is encoded in the spectral properties of the CME operator. Markov State Models (MSMs) provide a general framework for analyzing and visualizing stochastic multistability and state transitions based on these spectral properties. Until now, however, this approach is either limited to low-dimensional systems or requires the use of high-performance computing facilities, thus limiting its usability.
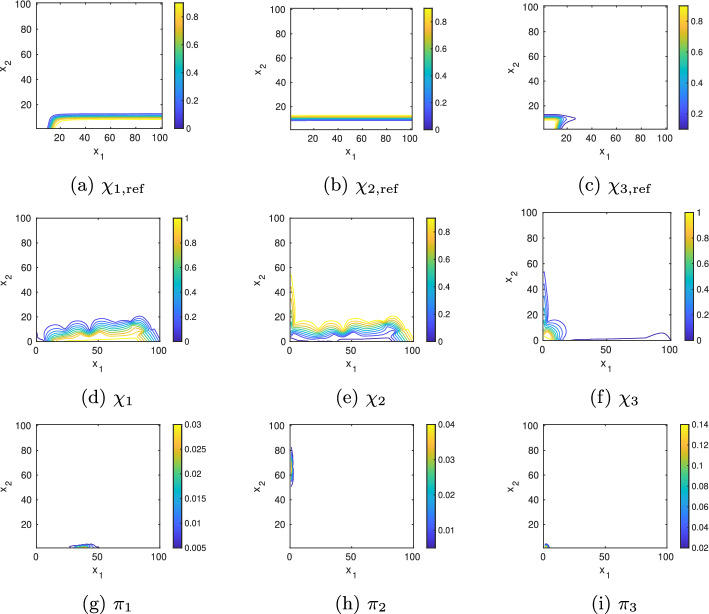
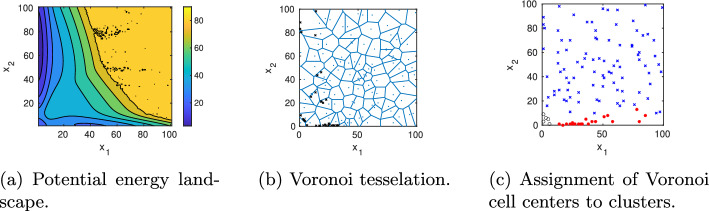
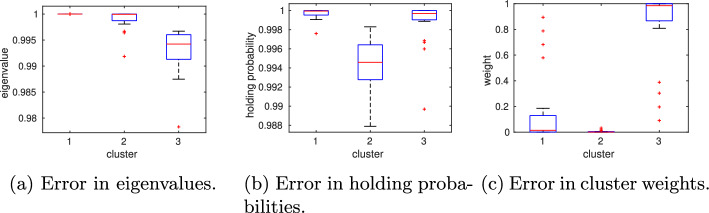
Results: We present a domain decomposition approach (DDA) that approximates the CME by a stochastic rate matrix on a discretized state space and projects the multistable dynamics to a lower dimensional MSM. To approximate the CME, we decompose the state space via a Voronoi tessellation and estimate transition probabilities by using adaptive sampling strategies. We apply the robust Perron cluster analysis (PCCA+) to construct the final MSM. Measures for uncertainty quantification are incorporated. As a proof of concept, we run the algorithm on a single PC and apply it to two GRN models, one for the genetic toggle switch and one describing macrophage polarization. By comparing the results with reference solutions, we demonstrate that our approach correctly identifies the number and location of metastable phenotypes with adequate accuracy and uncertainty bounds. We show that accuracy mainly depends on the total number of Voronoi cells, whereas uncertainty is determined by the number of sampling points.
Conclusions: A DDA enables the efficient computation of MSMs with quantified uncertainty. Since the algorithm is trivially parallelizable, it can be applied to larger systems, which will inevitably lead to new insights into cell-regulatory dynamics.
期刊介绍:
BMC Bioinformatics is an open access, peer-reviewed journal that considers articles on all aspects of the development, testing and novel application of computational and statistical methods for the modeling and analysis of all kinds of biological data, as well as other areas of computational biology.
BMC Bioinformatics is part of the BMC series which publishes subject-specific journals focused on the needs of individual research communities across all areas of biology and medicine. We offer an efficient, fair and friendly peer review service, and are committed to publishing all sound science, provided that there is some advance in knowledge presented by the work.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


