Zaafir M. Dulloo, Ion Ghiviriga, Mary E. Law, Sarvesh K. Verma, Abhisheak Sharma, Brian K. Law and Ronald K. Castellano
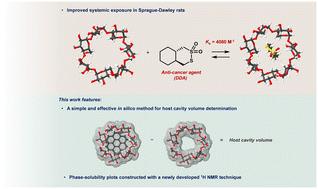
{"title":"用于改善全身暴露的二硫键破坏剂的β -环糊精配方。","authors":"Zaafir M. Dulloo, Ion Ghiviriga, Mary E. Law, Sarvesh K. Verma, Abhisheak Sharma, Brian K. Law and Ronald K. Castellano","doi":"10.1039/D5MD00334B","DOIUrl":null,"url":null,"abstract":"<p >Disulfide-bond disrupting agents (DDAs) are a class of cyclic thiosulfonates that have been shown to kill human epidermal growth factor receptor (HER) family-overexpressing breast cancer (BC) cells selectively and with no adverse side effects. Previous structure–activity relationships suggested a strong correlation between DDA lipophilicity and potency. In this study, we present the use of cyclodextrins (CDs) as molecular excipients to address the possible solubilizing drawback of increasingly lipophilic DDAs in oral administrations. The formulation of tcyDTDO, a potent second-generation DDA, with beta-cyclodextrin (BCD) and 2-hydroxypropyl-beta-cyclodextrin (HPB) was investigated. The choice of BCD as an optimal host over other CDs was guided by two <em>in silico</em> methods, namely: (1) host cavity volume estimations using a computational modeling approach and (2) binding energy (BE) calculations from simulations of different complexation geometries. A solid-state inclusion complex (IC) between tcyDTDO and BCD was prepared by kneading. Characterization by ATR-FTIR revealed positioning of tcyDTDO inside the cavity of BCD. Phase-solubility plots were constructed using NMR spectroscopy to measure the concentrations of host and guest in solution; a powerful technique that has yet to be exploited in the context of host–guest chemistry. The <em>A</em><small><sub>L</sub></small>-type plots obtained pointed to the formation of 1 : 1 complexes with both BCD and HPB. BCD formed a stronger complex with tcyDTDO (<em>K</em><small><sub>a</sub></small> of 4090 M<small><sup>−1</sup></small>) although the solubility of tcyDTDO was enhanced by only 3-fold from an intrinsic solubility of 1.58 mM. Contrastingly, HPB displayed a lower affinity for tcyDTDO (<em>K</em><small><sub>a</sub></small> of 81 M<small><sup>−1</sup></small>) but resulted in a remarkable 90-fold increase in solubility with tcyDTDO concentrations approaching 150 mM. Encapsulation of tcyDTDO in both cases did not hinder its anti-cancer activity as they retained cytotoxicity against MDA-MB-468 (EGFR+) BC cells <em>in vitro</em>. More striking was the superior pharmacokinetic profile and systemic exposure of tcyDTDO observed in male Sprague-Dawley rats when formulated with BCD as indicated by an area under the concentration <em>vs.</em> time curve (AUC<small><sub>0–24</sub></small>) of 3150 ± 381 ng h mL<small><sup>−1</sup></small>. This work suggests a correlation between <em>K</em><small><sub>a</sub></small> and <em>in vivo</em> pharmacokinetics of DDAs following their complexation with CDs and provides an ameliorated approach for their oral administration in future animal studies.</p>","PeriodicalId":21462,"journal":{"name":"RSC medicinal chemistry","volume":" 8","pages":" 3622-3632"},"PeriodicalIF":3.6000,"publicationDate":"2025-05-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12124217/pdf/","citationCount":"0","resultStr":"{\"title\":\"Beta-cyclodextrin formulation of a disulfide-bond disrupting agent for improved systemic exposure†\",\"authors\":\"Zaafir M. Dulloo, Ion Ghiviriga, Mary E. Law, Sarvesh K. Verma, Abhisheak Sharma, Brian K. Law and Ronald K. Castellano\",\"doi\":\"10.1039/D5MD00334B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Disulfide-bond disrupting agents (DDAs) are a class of cyclic thiosulfonates that have been shown to kill human epidermal growth factor receptor (HER) family-overexpressing breast cancer (BC) cells selectively and with no adverse side effects. Previous structure–activity relationships suggested a strong correlation between DDA lipophilicity and potency. In this study, we present the use of cyclodextrins (CDs) as molecular excipients to address the possible solubilizing drawback of increasingly lipophilic DDAs in oral administrations. The formulation of tcyDTDO, a potent second-generation DDA, with beta-cyclodextrin (BCD) and 2-hydroxypropyl-beta-cyclodextrin (HPB) was investigated. The choice of BCD as an optimal host over other CDs was guided by two <em>in silico</em> methods, namely: (1) host cavity volume estimations using a computational modeling approach and (2) binding energy (BE) calculations from simulations of different complexation geometries. A solid-state inclusion complex (IC) between tcyDTDO and BCD was prepared by kneading. Characterization by ATR-FTIR revealed positioning of tcyDTDO inside the cavity of BCD. Phase-solubility plots were constructed using NMR spectroscopy to measure the concentrations of host and guest in solution; a powerful technique that has yet to be exploited in the context of host–guest chemistry. The <em>A</em><small><sub>L</sub></small>-type plots obtained pointed to the formation of 1 : 1 complexes with both BCD and HPB. BCD formed a stronger complex with tcyDTDO (<em>K</em><small><sub>a</sub></small> of 4090 M<small><sup>−1</sup></small>) although the solubility of tcyDTDO was enhanced by only 3-fold from an intrinsic solubility of 1.58 mM. Contrastingly, HPB displayed a lower affinity for tcyDTDO (<em>K</em><small><sub>a</sub></small> of 81 M<small><sup>−1</sup></small>) but resulted in a remarkable 90-fold increase in solubility with tcyDTDO concentrations approaching 150 mM. Encapsulation of tcyDTDO in both cases did not hinder its anti-cancer activity as they retained cytotoxicity against MDA-MB-468 (EGFR+) BC cells <em>in vitro</em>. More striking was the superior pharmacokinetic profile and systemic exposure of tcyDTDO observed in male Sprague-Dawley rats when formulated with BCD as indicated by an area under the concentration <em>vs.</em> time curve (AUC<small><sub>0–24</sub></small>) of 3150 ± 381 ng h mL<small><sup>−1</sup></small>. This work suggests a correlation between <em>K</em><small><sub>a</sub></small> and <em>in vivo</em> pharmacokinetics of DDAs following their complexation with CDs and provides an ameliorated approach for their oral administration in future animal studies.</p>\",\"PeriodicalId\":21462,\"journal\":{\"name\":\"RSC medicinal chemistry\",\"volume\":\" 8\",\"pages\":\" 3622-3632\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2025-05-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12124217/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"RSC medicinal chemistry\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/md/d5md00334b\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"RSC medicinal chemistry","FirstCategoryId":"3","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/md/d5md00334b","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
二硫键破坏剂(DDAs)是一类环硫代磺酸盐,已被证明可选择性地杀死人表皮生长因子受体(HER)家族过表达的乳腺癌(BC)细胞,且无不良副作用。先前的构效关系表明,DDA的亲脂性与效价之间存在很强的相关性。在这项研究中,我们提出使用环糊精(CDs)作为分子赋形剂,以解决口服给药中日益亲脂性dda可能的溶解缺陷。研究了以β -环糊精(BCD)和2-羟丙基- β -环糊精(HPB)为主要原料的二代强效DDA - tcyDTDO的合成。BCD作为最佳宿主的选择是由两种计算机方法指导的,即:(1)使用计算建模方法估计宿主腔体积,(2)通过模拟不同络合几何形状计算结合能(BE)。采用捏合法制备了tcyDTDO和BCD之间的固体包合物(IC)。ATR-FTIR表征揭示了tcyDTDO在BCD腔内的定位。采用核磁共振谱法构建相溶解度图,测定溶液中主客体的浓度;这是一种强大的技术,但在主客体化学的背景下尚未被利用。得到的A - l型图表明BCD和HPB都形成了1:1的配合物。BCD与tcyDTDO形成了更强的配合物(K a为4090 M-1),尽管tcyDTDO的溶解度仅从1.58 mM的固有溶解度提高了3倍。HPB对tcyDTDO的亲和力较低(K a为81 M-1),但当tcyDTDO浓度接近150 mM时,其溶解度显著增加90倍。在这两种情况下,tcyDTDO的包封都不会阻碍其抗癌活性,因为它们在体外对MDA-MB-468 (EGFR+) BC细胞保持细胞毒性。更引人注目的是,当与BCD配制时,雄性Sprague-Dawley大鼠的tcyDTDO具有优异的药代动力学特征和全身暴露,浓度与时间曲线(AUC0-24)下的面积为3150±381 ng h mL-1。本研究提示了ka与dda与cd络合后的体内药代动力学之间的相关性,并为其在未来动物研究中的口服给药提供了改进的方法。
Beta-cyclodextrin formulation of a disulfide-bond disrupting agent for improved systemic exposure†
Disulfide-bond disrupting agents (DDAs) are a class of cyclic thiosulfonates that have been shown to kill human epidermal growth factor receptor (HER) family-overexpressing breast cancer (BC) cells selectively and with no adverse side effects. Previous structure–activity relationships suggested a strong correlation between DDA lipophilicity and potency. In this study, we present the use of cyclodextrins (CDs) as molecular excipients to address the possible solubilizing drawback of increasingly lipophilic DDAs in oral administrations. The formulation of tcyDTDO, a potent second-generation DDA, with beta-cyclodextrin (BCD) and 2-hydroxypropyl-beta-cyclodextrin (HPB) was investigated. The choice of BCD as an optimal host over other CDs was guided by two in silico methods, namely: (1) host cavity volume estimations using a computational modeling approach and (2) binding energy (BE) calculations from simulations of different complexation geometries. A solid-state inclusion complex (IC) between tcyDTDO and BCD was prepared by kneading. Characterization by ATR-FTIR revealed positioning of tcyDTDO inside the cavity of BCD. Phase-solubility plots were constructed using NMR spectroscopy to measure the concentrations of host and guest in solution; a powerful technique that has yet to be exploited in the context of host–guest chemistry. The AL-type plots obtained pointed to the formation of 1 : 1 complexes with both BCD and HPB. BCD formed a stronger complex with tcyDTDO (Ka of 4090 M−1) although the solubility of tcyDTDO was enhanced by only 3-fold from an intrinsic solubility of 1.58 mM. Contrastingly, HPB displayed a lower affinity for tcyDTDO (Ka of 81 M−1) but resulted in a remarkable 90-fold increase in solubility with tcyDTDO concentrations approaching 150 mM. Encapsulation of tcyDTDO in both cases did not hinder its anti-cancer activity as they retained cytotoxicity against MDA-MB-468 (EGFR+) BC cells in vitro. More striking was the superior pharmacokinetic profile and systemic exposure of tcyDTDO observed in male Sprague-Dawley rats when formulated with BCD as indicated by an area under the concentration vs. time curve (AUC0–24) of 3150 ± 381 ng h mL−1. This work suggests a correlation between Ka and in vivo pharmacokinetics of DDAs following their complexation with CDs and provides an ameliorated approach for their oral administration in future animal studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


