{"title":"残基保存和溶剂可及性(几乎)是预测蛋白质突变效应所需的全部。","authors":"Matsvei Tsishyn, Pauline Hermans, Marianne Rooman, Fabrizio Pucci","doi":"10.1093/bioinformatics/btaf322","DOIUrl":null,"url":null,"abstract":"<p><strong>Motivation: </strong>Predicting how mutations impact protein biophysical properties remains a significant challenge in computational biology. In recent years, numerous predictors, primarily deep learning models, have been developed to address this problem; however, issues such as their lack of interpretability and limited accuracy persist.</p><p><strong>Results: </strong>We showed that a simple evolutionary score, based on the log-odd ratio of wild-type and mutated residue frequencies in evolutionary related proteins, when scaled by the residue's relative solvent accessibility, performs on par with or slightly outperforms most of the benchmarked predictors, many of which are considerably more complex. The evaluation is performed on mutations from the ProteinGym deep mutational scanning dataset collection, which measures various properties such as stability, activity or fitness. This raises further questions about what these complex models actually learn and highlights their limitations in addressing prediction of mutational landscape.</p><p><strong>Availability and implementation: </strong>The RSALOR model is available as a user-friendly Python package that can be installed from the PyPI repository. The code is freely available at https://github.com/3BioCompBio/RSALOR.</p>","PeriodicalId":93899,"journal":{"name":"Bioinformatics (Oxford, England)","volume":" ","pages":""},"PeriodicalIF":5.4000,"publicationDate":"2025-06-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12202862/pdf/","citationCount":"0","resultStr":"{\"title\":\"Residue conservation and solvent accessibility are (almost) all you need for predicting mutational effects in proteins.\",\"authors\":\"Matsvei Tsishyn, Pauline Hermans, Marianne Rooman, Fabrizio Pucci\",\"doi\":\"10.1093/bioinformatics/btaf322\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Motivation: </strong>Predicting how mutations impact protein biophysical properties remains a significant challenge in computational biology. In recent years, numerous predictors, primarily deep learning models, have been developed to address this problem; however, issues such as their lack of interpretability and limited accuracy persist.</p><p><strong>Results: </strong>We showed that a simple evolutionary score, based on the log-odd ratio of wild-type and mutated residue frequencies in evolutionary related proteins, when scaled by the residue's relative solvent accessibility, performs on par with or slightly outperforms most of the benchmarked predictors, many of which are considerably more complex. The evaluation is performed on mutations from the ProteinGym deep mutational scanning dataset collection, which measures various properties such as stability, activity or fitness. This raises further questions about what these complex models actually learn and highlights their limitations in addressing prediction of mutational landscape.</p><p><strong>Availability and implementation: </strong>The RSALOR model is available as a user-friendly Python package that can be installed from the PyPI repository. The code is freely available at https://github.com/3BioCompBio/RSALOR.</p>\",\"PeriodicalId\":93899,\"journal\":{\"name\":\"Bioinformatics (Oxford, England)\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2025-06-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12202862/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics (Oxford, England)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1093/bioinformatics/btaf322\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics (Oxford, England)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/bioinformatics/btaf322","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Residue conservation and solvent accessibility are (almost) all you need for predicting mutational effects in proteins.
Motivation: Predicting how mutations impact protein biophysical properties remains a significant challenge in computational biology. In recent years, numerous predictors, primarily deep learning models, have been developed to address this problem; however, issues such as their lack of interpretability and limited accuracy persist.
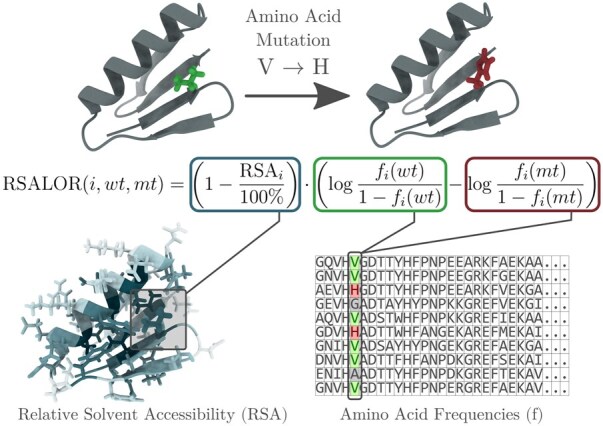
Results: We showed that a simple evolutionary score, based on the log-odd ratio of wild-type and mutated residue frequencies in evolutionary related proteins, when scaled by the residue's relative solvent accessibility, performs on par with or slightly outperforms most of the benchmarked predictors, many of which are considerably more complex. The evaluation is performed on mutations from the ProteinGym deep mutational scanning dataset collection, which measures various properties such as stability, activity or fitness. This raises further questions about what these complex models actually learn and highlights their limitations in addressing prediction of mutational landscape.
Availability and implementation: The RSALOR model is available as a user-friendly Python package that can be installed from the PyPI repository. The code is freely available at https://github.com/3BioCompBio/RSALOR.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


