{"title":"通过MD模拟探索多巴胺D2受体配体特异性激活动力学","authors":"Samman Mansoor, and , Giulia Morra*, ","doi":"10.1021/acs.jcim.4c0210110.1021/acs.jcim.4c02101","DOIUrl":null,"url":null,"abstract":"<p >The human G protein-coupled receptor (GPCR) dopamine 2 receptor (D<sub>2</sub>R) is an essential target of antipsychotic drugs. The modulation of downstream GPCR signaling induced by different agonists, termed functional selectivity, has potentially a great impact on drug discovery and control of side effects. The molecular origin of this modulation is, however, not fully understood. Here, the structural determinants underlying the response of D<sub>2</sub>R to binding of the endogenous agonist dopamine, the partial agonist aripiprazole, and the antagonist sulpiride are investigated at full atomistic resolution by molecular dynamics simulations. Multiple replicas covering 18 μs per system allow us to model binding mode and long-range effects of ligands and specifically modulation at the transmembrane helices (TMs) and at the intracellular interface. The dopamine-bound complex maintains the interaction points on TM3, TM5, and TM6 that lead to partial opening of the ionic lock required for the outward movement of TM6, whereas the binding mode of sulpiride disrupts the toggle switch, thereby globally altering the TM dynamics, which is conserved in the other two ligands. Moreover, we predict a significant impact of the partial agonist aripiprazole both on the extracellular loop EL2, on the N-terminal conformation of TM5, and on the dynamics of TM4, which in turn induces the perturbation of intracellular loop IL2 at the intracellular side. The latter loses its helical conformation, leading to a structure that is not competent for arrestin binding in a protein–protein docking computational experiment. These structural changes are accompanied by a modulation of cholesterol interactions with the receptor. Our model suggests that aripiprazole might cause poor arrestin recruitment by modulating TM4 and TM5 down to the intracellular side through the destabilization of a hydrophobic binding pocket at TM5, in agreement with previous mutational data.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 10","pages":"5050–5061 5050–5061"},"PeriodicalIF":5.3000,"publicationDate":"2025-05-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Insights into Ligand-Specific Activation Dynamics of Dopamine D2 Receptor Explored by MD Simulations\",\"authors\":\"Samman Mansoor, and , Giulia Morra*, \",\"doi\":\"10.1021/acs.jcim.4c0210110.1021/acs.jcim.4c02101\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The human G protein-coupled receptor (GPCR) dopamine 2 receptor (D<sub>2</sub>R) is an essential target of antipsychotic drugs. The modulation of downstream GPCR signaling induced by different agonists, termed functional selectivity, has potentially a great impact on drug discovery and control of side effects. The molecular origin of this modulation is, however, not fully understood. Here, the structural determinants underlying the response of D<sub>2</sub>R to binding of the endogenous agonist dopamine, the partial agonist aripiprazole, and the antagonist sulpiride are investigated at full atomistic resolution by molecular dynamics simulations. Multiple replicas covering 18 μs per system allow us to model binding mode and long-range effects of ligands and specifically modulation at the transmembrane helices (TMs) and at the intracellular interface. The dopamine-bound complex maintains the interaction points on TM3, TM5, and TM6 that lead to partial opening of the ionic lock required for the outward movement of TM6, whereas the binding mode of sulpiride disrupts the toggle switch, thereby globally altering the TM dynamics, which is conserved in the other two ligands. Moreover, we predict a significant impact of the partial agonist aripiprazole both on the extracellular loop EL2, on the N-terminal conformation of TM5, and on the dynamics of TM4, which in turn induces the perturbation of intracellular loop IL2 at the intracellular side. The latter loses its helical conformation, leading to a structure that is not competent for arrestin binding in a protein–protein docking computational experiment. These structural changes are accompanied by a modulation of cholesterol interactions with the receptor. Our model suggests that aripiprazole might cause poor arrestin recruitment by modulating TM4 and TM5 down to the intracellular side through the destabilization of a hydrophobic binding pocket at TM5, in agreement with previous mutational data.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 10\",\"pages\":\"5050–5061 5050–5061\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-05-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.4c02101\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.4c02101","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Insights into Ligand-Specific Activation Dynamics of Dopamine D2 Receptor Explored by MD Simulations
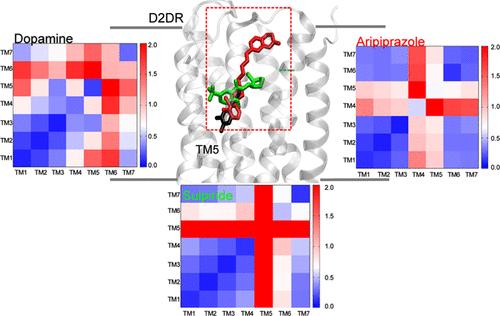
The human G protein-coupled receptor (GPCR) dopamine 2 receptor (D2R) is an essential target of antipsychotic drugs. The modulation of downstream GPCR signaling induced by different agonists, termed functional selectivity, has potentially a great impact on drug discovery and control of side effects. The molecular origin of this modulation is, however, not fully understood. Here, the structural determinants underlying the response of D2R to binding of the endogenous agonist dopamine, the partial agonist aripiprazole, and the antagonist sulpiride are investigated at full atomistic resolution by molecular dynamics simulations. Multiple replicas covering 18 μs per system allow us to model binding mode and long-range effects of ligands and specifically modulation at the transmembrane helices (TMs) and at the intracellular interface. The dopamine-bound complex maintains the interaction points on TM3, TM5, and TM6 that lead to partial opening of the ionic lock required for the outward movement of TM6, whereas the binding mode of sulpiride disrupts the toggle switch, thereby globally altering the TM dynamics, which is conserved in the other two ligands. Moreover, we predict a significant impact of the partial agonist aripiprazole both on the extracellular loop EL2, on the N-terminal conformation of TM5, and on the dynamics of TM4, which in turn induces the perturbation of intracellular loop IL2 at the intracellular side. The latter loses its helical conformation, leading to a structure that is not competent for arrestin binding in a protein–protein docking computational experiment. These structural changes are accompanied by a modulation of cholesterol interactions with the receptor. Our model suggests that aripiprazole might cause poor arrestin recruitment by modulating TM4 and TM5 down to the intracellular side through the destabilization of a hydrophobic binding pocket at TM5, in agreement with previous mutational data.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


