Babken G. Beglaryan, Aleksandr S. Zakuskin, Viktor A. Nemchenko and Timur A. Labutin*,
{"title":"迈向准确的多环芳烃红外光谱预测:处理电荷效应与经典和深度学习模型","authors":"Babken G. Beglaryan, Aleksandr S. Zakuskin, Viktor A. Nemchenko and Timur A. Labutin*, ","doi":"10.1021/acs.jcim.5c0037210.1021/acs.jcim.5c00372","DOIUrl":null,"url":null,"abstract":"<p >Polycyclic aromatic hydrocarbons (PAHs) play a crucial role in astrochemistry, environmental studies, and combustion chemistry, yet interpreting their infrared (IR) spectra remains challenging due to the similarity of spectral features of many molecules. The presumable presence of both neutral and charged PAHs in mixtures complicates spectra interpretation, too. While first-principle calculations provide accurate spectral predictions, their high computational cost limits scalability. This study employs machine learning (ML) to predict PAH IR spectra, emphasizing the applicability of the developed models simultaneously for neutral and ionized molecules. Two models are introduced: an XGBoost model trained on Morgan fingerprints and a graph neural network (GNN) that employs molecular graph representations. Molecular charges are treated by incorporating their one-hot or learnable NN encodings to molecular representations. Both models demonstrate excellent predictive capabilities, for the first time enabling fast and accurate prediction of charged PAHs IR spectra. While the XGBoost model demonstrates the highest accuracy achieved to date, the GNN shows significant promise for future advancements due to the inherent capabilities of molecular graph representations. Remaining challenges, such as the scarcity of data on heteroatomic PAHs, and potential approaches of addressing them are also discussed in the manuscript.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 10","pages":"4854–4865 4854–4865"},"PeriodicalIF":5.3000,"publicationDate":"2025-05-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Toward Accurate PAH IR Spectra Prediction: Handling Charge Effects with Classical and Deep Learning Models\",\"authors\":\"Babken G. Beglaryan, Aleksandr S. Zakuskin, Viktor A. Nemchenko and Timur A. Labutin*, \",\"doi\":\"10.1021/acs.jcim.5c0037210.1021/acs.jcim.5c00372\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Polycyclic aromatic hydrocarbons (PAHs) play a crucial role in astrochemistry, environmental studies, and combustion chemistry, yet interpreting their infrared (IR) spectra remains challenging due to the similarity of spectral features of many molecules. The presumable presence of both neutral and charged PAHs in mixtures complicates spectra interpretation, too. While first-principle calculations provide accurate spectral predictions, their high computational cost limits scalability. This study employs machine learning (ML) to predict PAH IR spectra, emphasizing the applicability of the developed models simultaneously for neutral and ionized molecules. Two models are introduced: an XGBoost model trained on Morgan fingerprints and a graph neural network (GNN) that employs molecular graph representations. Molecular charges are treated by incorporating their one-hot or learnable NN encodings to molecular representations. Both models demonstrate excellent predictive capabilities, for the first time enabling fast and accurate prediction of charged PAHs IR spectra. While the XGBoost model demonstrates the highest accuracy achieved to date, the GNN shows significant promise for future advancements due to the inherent capabilities of molecular graph representations. Remaining challenges, such as the scarcity of data on heteroatomic PAHs, and potential approaches of addressing them are also discussed in the manuscript.</p>\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"65 10\",\"pages\":\"4854–4865 4854–4865\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-05-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00372\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00372","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Toward Accurate PAH IR Spectra Prediction: Handling Charge Effects with Classical and Deep Learning Models
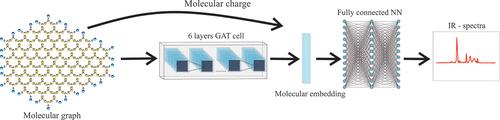
Polycyclic aromatic hydrocarbons (PAHs) play a crucial role in astrochemistry, environmental studies, and combustion chemistry, yet interpreting their infrared (IR) spectra remains challenging due to the similarity of spectral features of many molecules. The presumable presence of both neutral and charged PAHs in mixtures complicates spectra interpretation, too. While first-principle calculations provide accurate spectral predictions, their high computational cost limits scalability. This study employs machine learning (ML) to predict PAH IR spectra, emphasizing the applicability of the developed models simultaneously for neutral and ionized molecules. Two models are introduced: an XGBoost model trained on Morgan fingerprints and a graph neural network (GNN) that employs molecular graph representations. Molecular charges are treated by incorporating their one-hot or learnable NN encodings to molecular representations. Both models demonstrate excellent predictive capabilities, for the first time enabling fast and accurate prediction of charged PAHs IR spectra. While the XGBoost model demonstrates the highest accuracy achieved to date, the GNN shows significant promise for future advancements due to the inherent capabilities of molecular graph representations. Remaining challenges, such as the scarcity of data on heteroatomic PAHs, and potential approaches of addressing them are also discussed in the manuscript.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


