评价Soay羊野生群体qtl的区域遗传力定位方法。
IF 3.9
2区 生物学
Q2 ECOLOGY
引用次数: 0
摘要
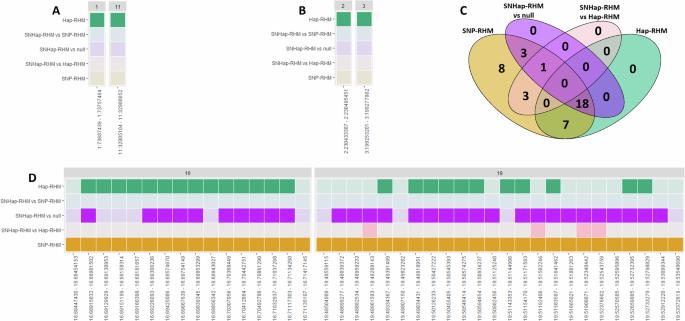
复杂性状及其遗传基础的研究对于理解形成自然种群的进化过程和机制至关重要。区域遗传作图(RHM)是一种估计基因组片段遗传力的方法,这些片段可能包含影响复杂性状的常见和罕见变异。这项研究很重要,因为它提高了我们检测导致表型变异的基因位点的能力,甚至是那些可能被传统方法(如全基因组关联研究(GWAS))遗漏的基因位点。在这里,我们比较了三种RHM方法:SNP-RHM,使用基于SNP基因型的基因组关系矩阵(GRMs);Hap-RHM,利用基于单倍型的grm;以及snp - rhm,它将基于snp的grm和基于单倍型的grm结合在一起。将这些方法应用于一个野生绵羊群体的数据,重点分析了11个多基因性状。将结果与先前GWAS的结果进行比较,以评估RHM在识别已知和新的相关位点方面的表现。我们发现,虽然区域矩阵的包含并不能解释与GWAS鉴定的性状变异相关的所有区域的显著变异,但它确实揭示了以前与性状变异无关的几个区域。这表明RHM方法可以为复杂性状的遗传结构提供额外的见解,突出GWAS单独可能忽略的基因组区域。这项研究强调了利用互补方法来充分了解自然种群中复杂性状的遗传基础的重要性。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Evaluating regional heritability mapping methods for identifying QTLs in a wild population of Soay sheep
The study of complex traits and their genetic underpinnings is crucial for understanding the evolutionary processes and mechanisms that shape natural populations. Regional heritability mapping (RHM) is a method for estimating the heritability of genomic segments that may contain both common and rare variants affecting a complex trait. This research is important because it advances our ability to detect genetic loci that contribute to phenotypic variation, even those that might be missed by traditional methods such as genome-wide association studies (GWAS). Here, we compare three RHM methods: SNP-RHM, which uses genomic relationship matrices (GRMs) based on SNP genotypes; Hap-RHM, which utilizes GRMs based on haplotypes; and SNHap-RHM, which integrates both SNP-based and haplotype-based GRMs jointly. These methods were applied to data from a wild population of sheep, focusing on the analysis of eleven polygenic traits. The results were compared with findings from previous GWAS to assess how RHM performed at identifying both known and novel associated loci. We found that while the inclusion of the regional matrix did not account for significant variation in all regions associated with trait variation as identified by GWAS, it did uncover several regions that were not previously linked to trait variation. This suggests that RHM methods can provide additional insights into the genetic architecture of complex traits, highlighting regions of the genome that may be overlooked by GWAS alone. This study underscores the importance of using complementary approaches to fully understand the genetic basis of complex traits in natural populations.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Heredity
生物-进化生物学
CiteScore
7.50
自引率
2.60%
发文量
84
审稿时长
4-8 weeks
期刊介绍:
Heredity is the official journal of the Genetics Society. It covers a broad range of topics within the field of genetics and therefore papers must address conceptual or applied issues of interest to the journal''s wide readership
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


