{"title":"1-吡唑啉脱氮的动力学研究:通过过渡态和二阶鞍区探索途径","authors":"Renuka Pradhan and Upakarasamy Lourderaj","doi":"10.1039/D5CP00798D","DOIUrl":null,"url":null,"abstract":"<p >The mechanism of 1-pyrazoline denitrogenation has garnered significant attention due to its remarkable stereoselectivity. In this study, the thermal denitrogenation mechanism of 1-pyrazoline was investigated using <em>ab initio</em> classical trajectory simulations to elucidate post-transition state and post-second-order saddle dynamics. Trajectories initiated from the synchronous transition state region predominantly followed the minimum energy pathway, forming the trimethylene diradical intermediate, which subsequently yielded cyclopropane with a preference for single inversion of the configuration. Additionally, the post-second-order saddle dynamics revealed that most trajectories followed the minimum energy path, offering alternative pathways for cyclopropane formation with retention of the configuration. In contrast, trajectories initiated from asynchronous transition state regions mostly deviated from the minimum energy path, leading to longer-lived diazenyl diradicals while still favoring single inversion in the final products. Despite significant diradical lifetimes, trajectories from all six transition-state regions exhibited a preference for single inversion cyclopropane formation, suggesting that product selectivity is dictated by dynamical effects rather than the reaction pathway.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 24","pages":" 12699-12710"},"PeriodicalIF":2.9000,"publicationDate":"2025-05-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Dynamical insights into denitrogenation of 1-pyrazoline: exploring pathways via transition states and a second-order saddle†\",\"authors\":\"Renuka Pradhan and Upakarasamy Lourderaj\",\"doi\":\"10.1039/D5CP00798D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The mechanism of 1-pyrazoline denitrogenation has garnered significant attention due to its remarkable stereoselectivity. In this study, the thermal denitrogenation mechanism of 1-pyrazoline was investigated using <em>ab initio</em> classical trajectory simulations to elucidate post-transition state and post-second-order saddle dynamics. Trajectories initiated from the synchronous transition state region predominantly followed the minimum energy pathway, forming the trimethylene diradical intermediate, which subsequently yielded cyclopropane with a preference for single inversion of the configuration. Additionally, the post-second-order saddle dynamics revealed that most trajectories followed the minimum energy path, offering alternative pathways for cyclopropane formation with retention of the configuration. In contrast, trajectories initiated from asynchronous transition state regions mostly deviated from the minimum energy path, leading to longer-lived diazenyl diradicals while still favoring single inversion in the final products. Despite significant diradical lifetimes, trajectories from all six transition-state regions exhibited a preference for single inversion cyclopropane formation, suggesting that product selectivity is dictated by dynamical effects rather than the reaction pathway.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 24\",\"pages\":\" 12699-12710\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-05-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00798d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00798d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Dynamical insights into denitrogenation of 1-pyrazoline: exploring pathways via transition states and a second-order saddle†
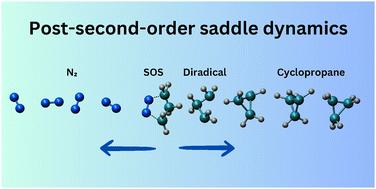
The mechanism of 1-pyrazoline denitrogenation has garnered significant attention due to its remarkable stereoselectivity. In this study, the thermal denitrogenation mechanism of 1-pyrazoline was investigated using ab initio classical trajectory simulations to elucidate post-transition state and post-second-order saddle dynamics. Trajectories initiated from the synchronous transition state region predominantly followed the minimum energy pathway, forming the trimethylene diradical intermediate, which subsequently yielded cyclopropane with a preference for single inversion of the configuration. Additionally, the post-second-order saddle dynamics revealed that most trajectories followed the minimum energy path, offering alternative pathways for cyclopropane formation with retention of the configuration. In contrast, trajectories initiated from asynchronous transition state regions mostly deviated from the minimum energy path, leading to longer-lived diazenyl diradicals while still favoring single inversion in the final products. Despite significant diradical lifetimes, trajectories from all six transition-state regions exhibited a preference for single inversion cyclopropane formation, suggesting that product selectivity is dictated by dynamical effects rather than the reaction pathway.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


