Neveen A Soliman, Mohamed A Elmonem, Ahmed F El-Sayed, Eman Ramadan, Ahmed M Badr, Fatma M Atia, Rasha Helmy, May O Amer, Ahmed Abd El-Raouf, Fadya M El-Garhy, Omnia M Abdel-Haseb, Tokka M Hassan, Yasmeen K Farouk, Ahmed El-Hosseiny, Usama Bakry, Asmaa Ali, Sheri Saleeb, Tasnim A Ghanim, Mahynour Albarbary, Ahmed Elmahy, Tarek Elnagdy, Amira Ragheb, Wael A Hassan, Ahmed Moustafa, Khaled Amer
{"title":"全基因组测序确定56.1%的早发性类固醇抵抗性肾病综合征家庭存在单基因疾病。","authors":"Neveen A Soliman, Mohamed A Elmonem, Ahmed F El-Sayed, Eman Ramadan, Ahmed M Badr, Fatma M Atia, Rasha Helmy, May O Amer, Ahmed Abd El-Raouf, Fadya M El-Garhy, Omnia M Abdel-Haseb, Tokka M Hassan, Yasmeen K Farouk, Ahmed El-Hosseiny, Usama Bakry, Asmaa Ali, Sheri Saleeb, Tasnim A Ghanim, Mahynour Albarbary, Ahmed Elmahy, Tarek Elnagdy, Amira Ragheb, Wael A Hassan, Ahmed Moustafa, Khaled Amer","doi":"10.1007/s00439-025-02752-y","DOIUrl":null,"url":null,"abstract":"<p><p>Genetic causes of steroid-resistant-nephrotic-syndrome (SRNS) represent a rapidly growing number of monogenic diseases. The reported diagnostic yield of various studies applying genetic panels and exome-sequencing to diagnose SRNS is usually < 30%. We performed genome-sequencing in a cohort of Egyptian SRNS patients. We recruited 47 SRNS patients belonging to 41 unrelated families [28 males/19 females; median (range): 6 (0.5-22 years)]. We established a pipeline for genome sequencing, bioinformatics analysis, variant curation and protein modeling at the Egypt Center for Research and Regenerative Medicine (ECRRM). Disease-causing variants were detected in 27/47 patients (57.4%) belonging to 23/41 families (56.1%), including nine novel variants in NPHS1, NPHS2, COL4A3, MYO1E, NUP93, PLCE1, PODXL, SMARCAL1 and WT1. Novel variants were confirmed by Sanger sequencing and were segregated in families of affected patients. NPHS2 was the most common causative gene in 8/23 (34.8%) of confirmed families, followed by NPHS1, WT1, and SMARCAL1 in 2/23 families (8.7%) each. All detected missense variants were evaluated through protein modeling and were predicted deleterious. Our study expanded the spectrum of SRNS disease-causing variants and revealed a monogenic cause in 56.1% of investigated families. In our cohort, no deep intronic or regulatory variants were detected by genome-sequencing. Pursuing genetic diagnosis in SRNS patients is crucial to inform clinical decision making, genetic counseling, transplantation strategy and prenatal diagnosis thus improving clinical outcome of affected patients.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"727-740"},"PeriodicalIF":3.6000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12222417/pdf/","citationCount":"0","resultStr":"{\"title\":\"Whole genome sequencing identifies monogenic disease in 56.1% of families with early-onset steroid-resistant nephrotic syndrome.\",\"authors\":\"Neveen A Soliman, Mohamed A Elmonem, Ahmed F El-Sayed, Eman Ramadan, Ahmed M Badr, Fatma M Atia, Rasha Helmy, May O Amer, Ahmed Abd El-Raouf, Fadya M El-Garhy, Omnia M Abdel-Haseb, Tokka M Hassan, Yasmeen K Farouk, Ahmed El-Hosseiny, Usama Bakry, Asmaa Ali, Sheri Saleeb, Tasnim A Ghanim, Mahynour Albarbary, Ahmed Elmahy, Tarek Elnagdy, Amira Ragheb, Wael A Hassan, Ahmed Moustafa, Khaled Amer\",\"doi\":\"10.1007/s00439-025-02752-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Genetic causes of steroid-resistant-nephrotic-syndrome (SRNS) represent a rapidly growing number of monogenic diseases. The reported diagnostic yield of various studies applying genetic panels and exome-sequencing to diagnose SRNS is usually < 30%. We performed genome-sequencing in a cohort of Egyptian SRNS patients. We recruited 47 SRNS patients belonging to 41 unrelated families [28 males/19 females; median (range): 6 (0.5-22 years)]. We established a pipeline for genome sequencing, bioinformatics analysis, variant curation and protein modeling at the Egypt Center for Research and Regenerative Medicine (ECRRM). Disease-causing variants were detected in 27/47 patients (57.4%) belonging to 23/41 families (56.1%), including nine novel variants in NPHS1, NPHS2, COL4A3, MYO1E, NUP93, PLCE1, PODXL, SMARCAL1 and WT1. Novel variants were confirmed by Sanger sequencing and were segregated in families of affected patients. NPHS2 was the most common causative gene in 8/23 (34.8%) of confirmed families, followed by NPHS1, WT1, and SMARCAL1 in 2/23 families (8.7%) each. All detected missense variants were evaluated through protein modeling and were predicted deleterious. Our study expanded the spectrum of SRNS disease-causing variants and revealed a monogenic cause in 56.1% of investigated families. In our cohort, no deep intronic or regulatory variants were detected by genome-sequencing. Pursuing genetic diagnosis in SRNS patients is crucial to inform clinical decision making, genetic counseling, transplantation strategy and prenatal diagnosis thus improving clinical outcome of affected patients.</p>\",\"PeriodicalId\":13175,\"journal\":{\"name\":\"Human Genetics\",\"volume\":\" \",\"pages\":\"727-740\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2025-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12222417/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00439-025-02752-y\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/5/22 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-025-02752-y","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/22 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Whole genome sequencing identifies monogenic disease in 56.1% of families with early-onset steroid-resistant nephrotic syndrome.
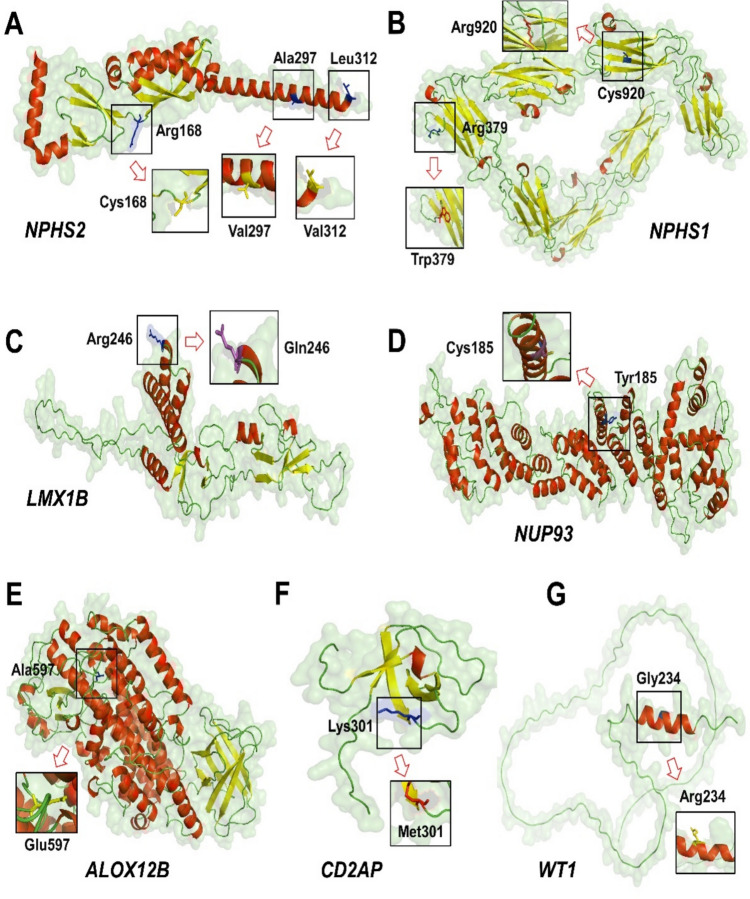
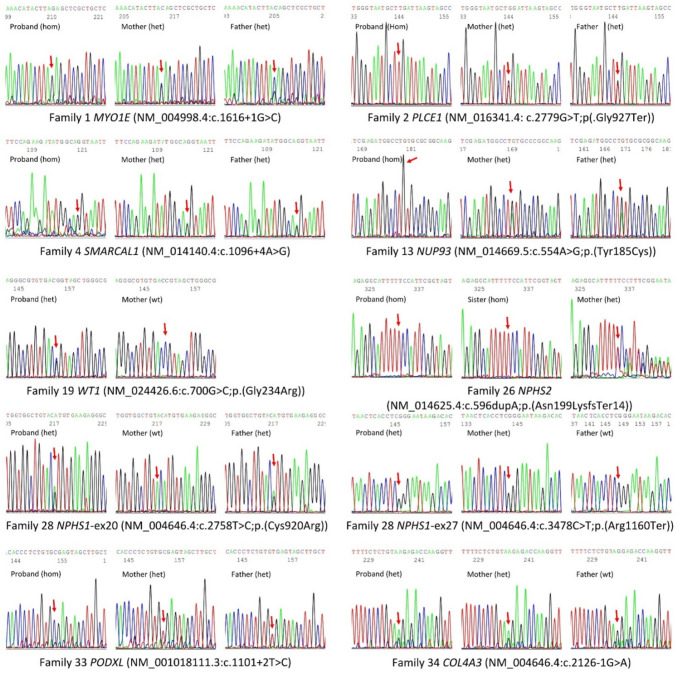
Genetic causes of steroid-resistant-nephrotic-syndrome (SRNS) represent a rapidly growing number of monogenic diseases. The reported diagnostic yield of various studies applying genetic panels and exome-sequencing to diagnose SRNS is usually < 30%. We performed genome-sequencing in a cohort of Egyptian SRNS patients. We recruited 47 SRNS patients belonging to 41 unrelated families [28 males/19 females; median (range): 6 (0.5-22 years)]. We established a pipeline for genome sequencing, bioinformatics analysis, variant curation and protein modeling at the Egypt Center for Research and Regenerative Medicine (ECRRM). Disease-causing variants were detected in 27/47 patients (57.4%) belonging to 23/41 families (56.1%), including nine novel variants in NPHS1, NPHS2, COL4A3, MYO1E, NUP93, PLCE1, PODXL, SMARCAL1 and WT1. Novel variants were confirmed by Sanger sequencing and were segregated in families of affected patients. NPHS2 was the most common causative gene in 8/23 (34.8%) of confirmed families, followed by NPHS1, WT1, and SMARCAL1 in 2/23 families (8.7%) each. All detected missense variants were evaluated through protein modeling and were predicted deleterious. Our study expanded the spectrum of SRNS disease-causing variants and revealed a monogenic cause in 56.1% of investigated families. In our cohort, no deep intronic or regulatory variants were detected by genome-sequencing. Pursuing genetic diagnosis in SRNS patients is crucial to inform clinical decision making, genetic counseling, transplantation strategy and prenatal diagnosis thus improving clinical outcome of affected patients.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


