Zain Aslam, Bibi Zubaida, Ranjha Khan, Mazhar Badshah, Muhammad Naeem
{"title":"在巴基斯坦一个患有严重和迅速进展的拉福拉病的近亲家庭中鉴定一种致病性NHLRC1变异。","authors":"Zain Aslam, Bibi Zubaida, Ranjha Khan, Mazhar Badshah, Muhammad Naeem","doi":"10.1186/s42494-024-00193-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Autosomal recessively inherited progressive myoclonic epilepsy of Lafora, which is also known as Lafora disease, is a fatal neurodegenerative disorder. It affects individuals in late childhood or early adolescence and presents with symptoms of progressive mental and physical deterioration. This disease is caused by pathogenic genetic variants in either of two genes: EPM2A on chromosome 6q24, encoding laforin, or NHLRC1 (EPM2B) on chromosome 6p22, encoding malin.</p><p><strong>Case presentation: </strong>In this study, we report a clinical and molecular investigation of Lafora disease segregating in a consanguineous Pakistani family. The proband presented with symptoms of rapidly progressive myoclonic epilepsy, but laboratory studies were negative for Lafora bodies and ragged red fibres. Sanger DNA sequencing of the genomic DNA of the proband for EPM2A and NHLRC1 identified a previously reported pathogenic nonsense variant in NHLRC1 (NM_198586.3:c.793C > T), which encodes the E3 ubiquitin ligase called malin. The NHLRC1 variant was found in a homozygous state in the proband, predicting a premature stop codon at position 265 (NP_940988.2:p.Arg265Ter). If the mRNA escaped nonsense-mediated decay, it would result in a truncated protein lacking 130 amino acids, including three NHL (NCL-1, HT2A, LIN-41) repeats.</p><p><strong>Conclusions: </strong>Our study emphasizes the role of molecular genetic testing in patients presenting with symptoms of progressive myoclonic epilepsy.</p>","PeriodicalId":33628,"journal":{"name":"Acta Epileptologica","volume":"7 1","pages":"7"},"PeriodicalIF":1.2000,"publicationDate":"2025-01-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11960279/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification of a pathogenic NHLRC1 variant in a consanguineous Pakistani family affected with severe and rapidly progressive Lafora disease.\",\"authors\":\"Zain Aslam, Bibi Zubaida, Ranjha Khan, Mazhar Badshah, Muhammad Naeem\",\"doi\":\"10.1186/s42494-024-00193-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Autosomal recessively inherited progressive myoclonic epilepsy of Lafora, which is also known as Lafora disease, is a fatal neurodegenerative disorder. It affects individuals in late childhood or early adolescence and presents with symptoms of progressive mental and physical deterioration. This disease is caused by pathogenic genetic variants in either of two genes: EPM2A on chromosome 6q24, encoding laforin, or NHLRC1 (EPM2B) on chromosome 6p22, encoding malin.</p><p><strong>Case presentation: </strong>In this study, we report a clinical and molecular investigation of Lafora disease segregating in a consanguineous Pakistani family. The proband presented with symptoms of rapidly progressive myoclonic epilepsy, but laboratory studies were negative for Lafora bodies and ragged red fibres. Sanger DNA sequencing of the genomic DNA of the proband for EPM2A and NHLRC1 identified a previously reported pathogenic nonsense variant in NHLRC1 (NM_198586.3:c.793C > T), which encodes the E3 ubiquitin ligase called malin. The NHLRC1 variant was found in a homozygous state in the proband, predicting a premature stop codon at position 265 (NP_940988.2:p.Arg265Ter). If the mRNA escaped nonsense-mediated decay, it would result in a truncated protein lacking 130 amino acids, including three NHL (NCL-1, HT2A, LIN-41) repeats.</p><p><strong>Conclusions: </strong>Our study emphasizes the role of molecular genetic testing in patients presenting with symptoms of progressive myoclonic epilepsy.</p>\",\"PeriodicalId\":33628,\"journal\":{\"name\":\"Acta Epileptologica\",\"volume\":\"7 1\",\"pages\":\"7\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2025-01-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11960279/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta Epileptologica\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s42494-024-00193-0\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CLINICAL NEUROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Epileptologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42494-024-00193-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
Identification of a pathogenic NHLRC1 variant in a consanguineous Pakistani family affected with severe and rapidly progressive Lafora disease.
Background: Autosomal recessively inherited progressive myoclonic epilepsy of Lafora, which is also known as Lafora disease, is a fatal neurodegenerative disorder. It affects individuals in late childhood or early adolescence and presents with symptoms of progressive mental and physical deterioration. This disease is caused by pathogenic genetic variants in either of two genes: EPM2A on chromosome 6q24, encoding laforin, or NHLRC1 (EPM2B) on chromosome 6p22, encoding malin.
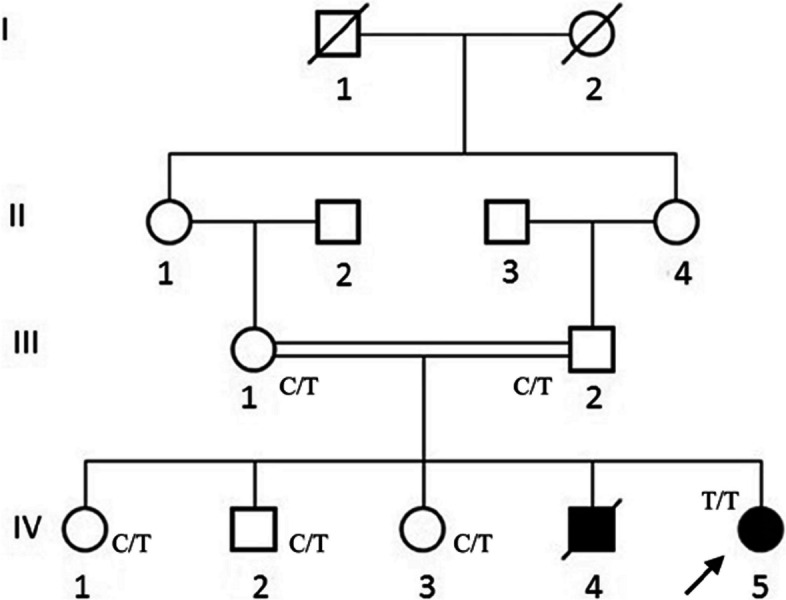
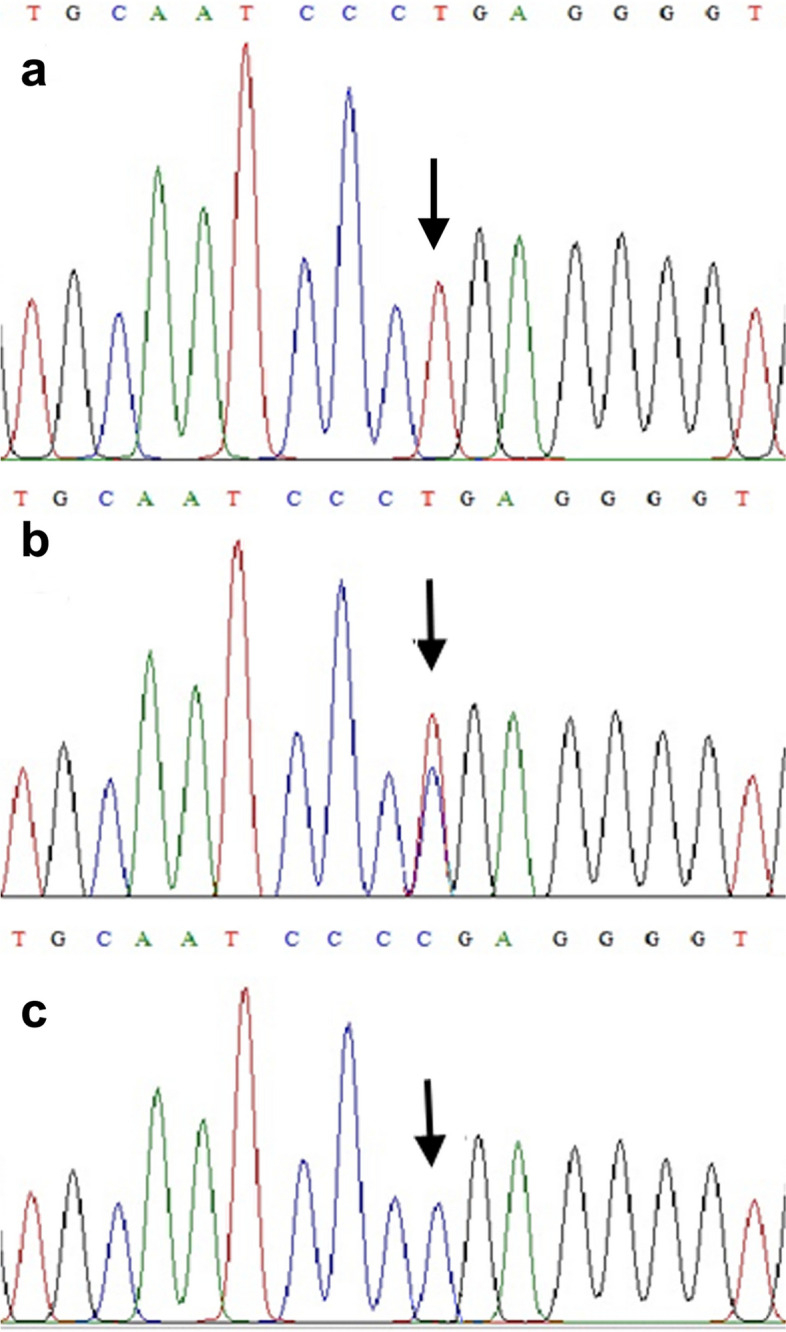
Case presentation: In this study, we report a clinical and molecular investigation of Lafora disease segregating in a consanguineous Pakistani family. The proband presented with symptoms of rapidly progressive myoclonic epilepsy, but laboratory studies were negative for Lafora bodies and ragged red fibres. Sanger DNA sequencing of the genomic DNA of the proband for EPM2A and NHLRC1 identified a previously reported pathogenic nonsense variant in NHLRC1 (NM_198586.3:c.793C > T), which encodes the E3 ubiquitin ligase called malin. The NHLRC1 variant was found in a homozygous state in the proband, predicting a premature stop codon at position 265 (NP_940988.2:p.Arg265Ter). If the mRNA escaped nonsense-mediated decay, it would result in a truncated protein lacking 130 amino acids, including three NHL (NCL-1, HT2A, LIN-41) repeats.
Conclusions: Our study emphasizes the role of molecular genetic testing in patients presenting with symptoms of progressive myoclonic epilepsy.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


