{"title":"成人尿素循环缺陷:具有潜在脑病基因编码的肝脏代谢紊乱的临床表现、诊断和治疗","authors":"Anibh Martin Das","doi":"10.1007/s11011-025-01619-5","DOIUrl":null,"url":null,"abstract":"<p><p>Hyperammonaemia is an important cause for encephalopathy. Ammonia is the waste product of amino acid degradation and cannot be excreted via urine. Ammonia is metabolized to water-soluble urea via the urea cycle. Hyperammonaemia not only occurs during acute liver failure, but also in rare genetically determined defects of enzymes or transporters involved in the urea cycle resulting in elevated ammonia concentrations. Enzyme defects include deficiency of carbamylphosphate synthase, N-acetylglutamate synthase, ornithine transcarbamylase, argininosuccinate lyase and arginase, transporter defects are citrin deficiency and HHH-syndrome. These urea cycle defects (UCD) mostly manifest for the first time during the neonatal period, infancy or childhood, however first clinical manifestations including encephalopathy may be observed in adulthood in milder forms. Therefore, physicians treating adults should be aware of clinical symptoms in UCD to make a timely diagnosis and initiate treatment. In adulthood, clinical symptoms are often uncharacteristic including headache, avoidance of high-protein food, psychiatric symptoms triggered by heavy exercise or delivery of a child, autism, attention deficit, lethargy, developmental delay and epilepsy. Elevated ammonia concentrations in blood are the biochemical hallmark. Some UCDs can be diagnosed at metabolite level, others only at genetic level. Treatment consists of eucaloric, low-protein diet supplemented with essential amino acids and vitamins/trace elements, and intake of arginine or citrulline. Pharmacological scavengers of nitrogen are benzoate and butyrate. If conservative therapy fails, hemodialysis should be considered. Prompt treatment during acute crises is essential for optimal outcome. Liver transplantation is considered in metabolically unstable patients. For arginase deficiency, enzyme replacement therapy is available.</p>","PeriodicalId":18685,"journal":{"name":"Metabolic brain disease","volume":"40 5","pages":"192"},"PeriodicalIF":3.5000,"publicationDate":"2025-04-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12033206/pdf/","citationCount":"0","resultStr":"{\"title\":\"Urea cycle defects in adulthood: clinical presentation, diagnosis and treatment in genetically encoded hepatic metabolic disorders with a potential for encephalopathy.\",\"authors\":\"Anibh Martin Das\",\"doi\":\"10.1007/s11011-025-01619-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hyperammonaemia is an important cause for encephalopathy. Ammonia is the waste product of amino acid degradation and cannot be excreted via urine. Ammonia is metabolized to water-soluble urea via the urea cycle. Hyperammonaemia not only occurs during acute liver failure, but also in rare genetically determined defects of enzymes or transporters involved in the urea cycle resulting in elevated ammonia concentrations. Enzyme defects include deficiency of carbamylphosphate synthase, N-acetylglutamate synthase, ornithine transcarbamylase, argininosuccinate lyase and arginase, transporter defects are citrin deficiency and HHH-syndrome. These urea cycle defects (UCD) mostly manifest for the first time during the neonatal period, infancy or childhood, however first clinical manifestations including encephalopathy may be observed in adulthood in milder forms. Therefore, physicians treating adults should be aware of clinical symptoms in UCD to make a timely diagnosis and initiate treatment. In adulthood, clinical symptoms are often uncharacteristic including headache, avoidance of high-protein food, psychiatric symptoms triggered by heavy exercise or delivery of a child, autism, attention deficit, lethargy, developmental delay and epilepsy. Elevated ammonia concentrations in blood are the biochemical hallmark. Some UCDs can be diagnosed at metabolite level, others only at genetic level. Treatment consists of eucaloric, low-protein diet supplemented with essential amino acids and vitamins/trace elements, and intake of arginine or citrulline. Pharmacological scavengers of nitrogen are benzoate and butyrate. If conservative therapy fails, hemodialysis should be considered. Prompt treatment during acute crises is essential for optimal outcome. Liver transplantation is considered in metabolically unstable patients. For arginase deficiency, enzyme replacement therapy is available.</p>\",\"PeriodicalId\":18685,\"journal\":{\"name\":\"Metabolic brain disease\",\"volume\":\"40 5\",\"pages\":\"192\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-04-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12033206/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Metabolic brain disease\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s11011-025-01619-5\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Metabolic brain disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11011-025-01619-5","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
Urea cycle defects in adulthood: clinical presentation, diagnosis and treatment in genetically encoded hepatic metabolic disorders with a potential for encephalopathy.
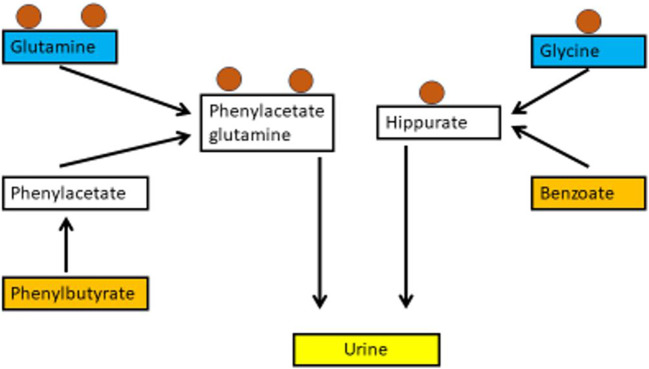
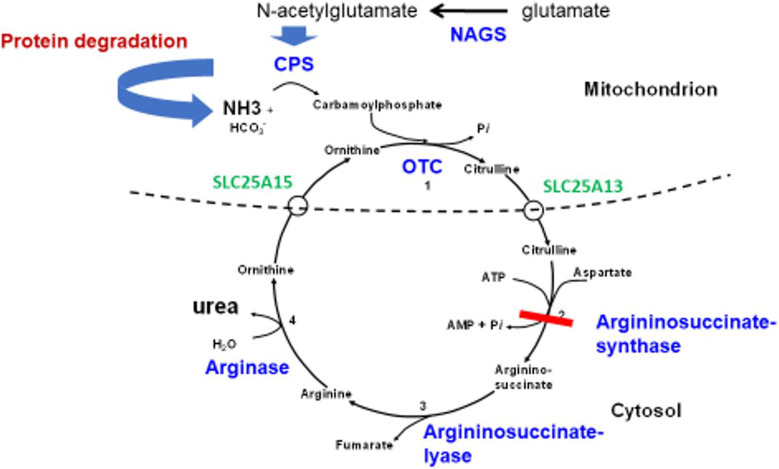
Hyperammonaemia is an important cause for encephalopathy. Ammonia is the waste product of amino acid degradation and cannot be excreted via urine. Ammonia is metabolized to water-soluble urea via the urea cycle. Hyperammonaemia not only occurs during acute liver failure, but also in rare genetically determined defects of enzymes or transporters involved in the urea cycle resulting in elevated ammonia concentrations. Enzyme defects include deficiency of carbamylphosphate synthase, N-acetylglutamate synthase, ornithine transcarbamylase, argininosuccinate lyase and arginase, transporter defects are citrin deficiency and HHH-syndrome. These urea cycle defects (UCD) mostly manifest for the first time during the neonatal period, infancy or childhood, however first clinical manifestations including encephalopathy may be observed in adulthood in milder forms. Therefore, physicians treating adults should be aware of clinical symptoms in UCD to make a timely diagnosis and initiate treatment. In adulthood, clinical symptoms are often uncharacteristic including headache, avoidance of high-protein food, psychiatric symptoms triggered by heavy exercise or delivery of a child, autism, attention deficit, lethargy, developmental delay and epilepsy. Elevated ammonia concentrations in blood are the biochemical hallmark. Some UCDs can be diagnosed at metabolite level, others only at genetic level. Treatment consists of eucaloric, low-protein diet supplemented with essential amino acids and vitamins/trace elements, and intake of arginine or citrulline. Pharmacological scavengers of nitrogen are benzoate and butyrate. If conservative therapy fails, hemodialysis should be considered. Prompt treatment during acute crises is essential for optimal outcome. Liver transplantation is considered in metabolically unstable patients. For arginase deficiency, enzyme replacement therapy is available.
期刊介绍:
Metabolic Brain Disease serves as a forum for the publication of outstanding basic and clinical papers on all metabolic brain disease, including both human and animal studies. The journal publishes papers on the fundamental pathogenesis of these disorders and on related experimental and clinical techniques and methodologies. Metabolic Brain Disease is directed to physicians, neuroscientists, internists, psychiatrists, neurologists, pathologists, and others involved in the research and treatment of a broad range of metabolic brain disorders.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


