José Miguel Brito Armas, Lucas Taoro-González, Elizabeth M C Fisher, Abraham Acevedo-Arozena
{"title":"TDP-43小鼠病理模型的挑战。","authors":"José Miguel Brito Armas, Lucas Taoro-González, Elizabeth M C Fisher, Abraham Acevedo-Arozena","doi":"10.1007/s00335-025-10131-1","DOIUrl":null,"url":null,"abstract":"<p><p>TDP-43 is a normally nuclear RNA binding protein that under pathological conditions may be excluded from the nucleus and deposited in the cytoplasm in the form of insoluble polyubiquitinated and polyphosphorylated inclusions. This nuclear exclusion coupled with cytoplasmic accumulation is called TDP-43 pathology and contributes to a range of disorders collectively known as TDP-43 proteinopathies. These include the great majority of amyotrophic lateral sclerosis (ALS) cases, all limbic-predominant age-related TDP-43 encephalopathy (LATE), as well as up to 50% of frontotemporal lobar degeneration (FTLD) and Alzheimer's disease (AD) cases. Thus, TDP-43 pathology is a common feature underlying a wide range of neurodegenerative conditions. However, modelling it has proven to be challenging, particularly generating models with concomitant TDP-43 loss of nuclear function and cytoplasmic inclusions. Here, focussing exclusively on mice, we discuss TDP-43 genetic models in terms of the presence of TDP-43 pathology, and we consider other models with TDP-43 pathology due to mutations in disparate genes. We also consider manipulations aimed at producing TDP-43 pathology, and we look at potential strategies to develop new, much needed models to address the many outstanding questions regarding how and why TDP-43 protein leaves the nucleus and accumulates in the cytoplasm, causing downstream dysfunction and devastating disease.</p>","PeriodicalId":18259,"journal":{"name":"Mammalian Genome","volume":" ","pages":"465-481"},"PeriodicalIF":2.7000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12130161/pdf/","citationCount":"0","resultStr":"{\"title\":\"Challenges of modelling TDP-43 pathology in mice.\",\"authors\":\"José Miguel Brito Armas, Lucas Taoro-González, Elizabeth M C Fisher, Abraham Acevedo-Arozena\",\"doi\":\"10.1007/s00335-025-10131-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>TDP-43 is a normally nuclear RNA binding protein that under pathological conditions may be excluded from the nucleus and deposited in the cytoplasm in the form of insoluble polyubiquitinated and polyphosphorylated inclusions. This nuclear exclusion coupled with cytoplasmic accumulation is called TDP-43 pathology and contributes to a range of disorders collectively known as TDP-43 proteinopathies. These include the great majority of amyotrophic lateral sclerosis (ALS) cases, all limbic-predominant age-related TDP-43 encephalopathy (LATE), as well as up to 50% of frontotemporal lobar degeneration (FTLD) and Alzheimer's disease (AD) cases. Thus, TDP-43 pathology is a common feature underlying a wide range of neurodegenerative conditions. However, modelling it has proven to be challenging, particularly generating models with concomitant TDP-43 loss of nuclear function and cytoplasmic inclusions. Here, focussing exclusively on mice, we discuss TDP-43 genetic models in terms of the presence of TDP-43 pathology, and we consider other models with TDP-43 pathology due to mutations in disparate genes. We also consider manipulations aimed at producing TDP-43 pathology, and we look at potential strategies to develop new, much needed models to address the many outstanding questions regarding how and why TDP-43 protein leaves the nucleus and accumulates in the cytoplasm, causing downstream dysfunction and devastating disease.</p>\",\"PeriodicalId\":18259,\"journal\":{\"name\":\"Mammalian Genome\",\"volume\":\" \",\"pages\":\"465-481\"},\"PeriodicalIF\":2.7000,\"publicationDate\":\"2025-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12130161/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Mammalian Genome\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00335-025-10131-1\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/4/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mammalian Genome","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00335-025-10131-1","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/29 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
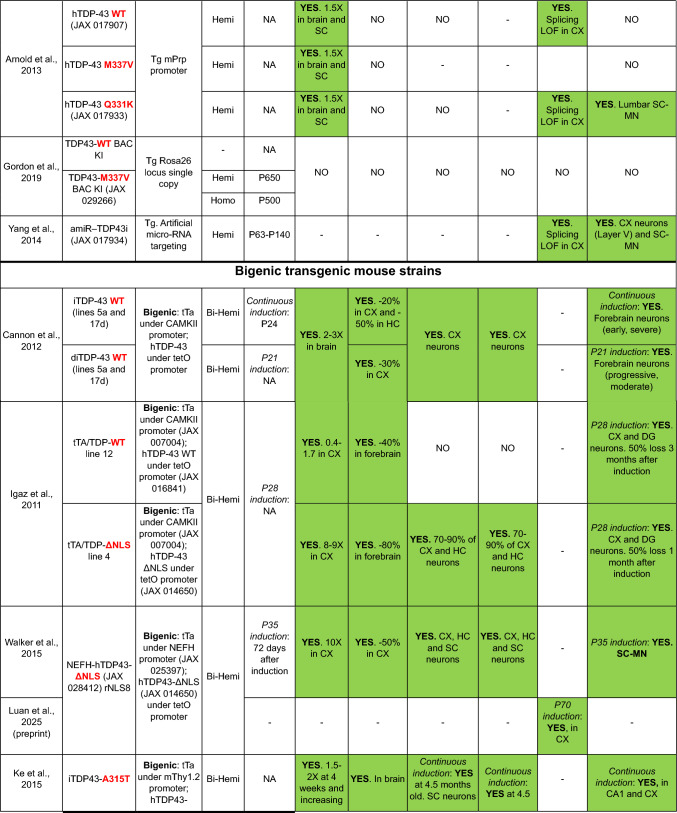
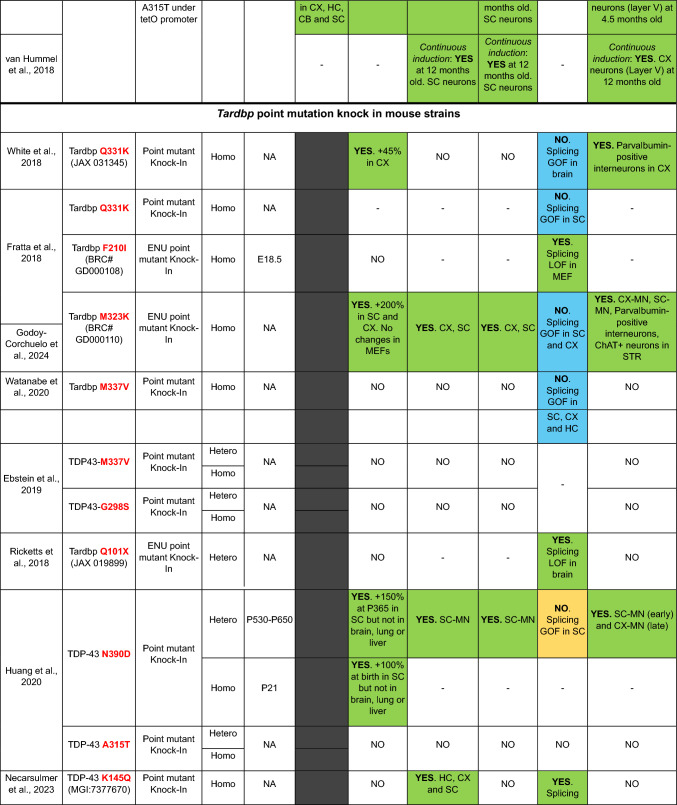
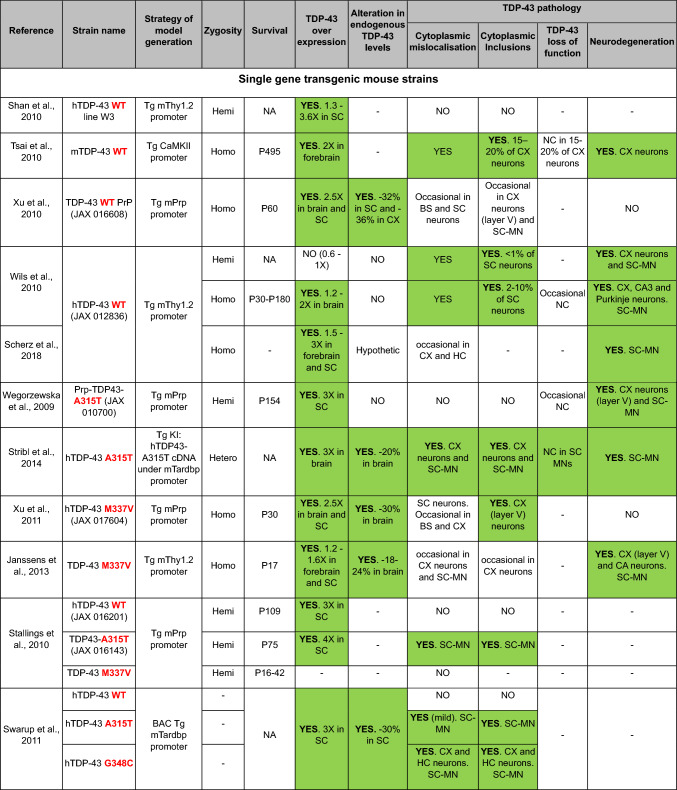
TDP-43 is a normally nuclear RNA binding protein that under pathological conditions may be excluded from the nucleus and deposited in the cytoplasm in the form of insoluble polyubiquitinated and polyphosphorylated inclusions. This nuclear exclusion coupled with cytoplasmic accumulation is called TDP-43 pathology and contributes to a range of disorders collectively known as TDP-43 proteinopathies. These include the great majority of amyotrophic lateral sclerosis (ALS) cases, all limbic-predominant age-related TDP-43 encephalopathy (LATE), as well as up to 50% of frontotemporal lobar degeneration (FTLD) and Alzheimer's disease (AD) cases. Thus, TDP-43 pathology is a common feature underlying a wide range of neurodegenerative conditions. However, modelling it has proven to be challenging, particularly generating models with concomitant TDP-43 loss of nuclear function and cytoplasmic inclusions. Here, focussing exclusively on mice, we discuss TDP-43 genetic models in terms of the presence of TDP-43 pathology, and we consider other models with TDP-43 pathology due to mutations in disparate genes. We also consider manipulations aimed at producing TDP-43 pathology, and we look at potential strategies to develop new, much needed models to address the many outstanding questions regarding how and why TDP-43 protein leaves the nucleus and accumulates in the cytoplasm, causing downstream dysfunction and devastating disease.
期刊介绍:
Mammalian Genome focuses on the experimental, theoretical and technical aspects of genetics, genomics, epigenetics and systems biology in mouse, human and other mammalian species, with an emphasis on the relationship between genotype and phenotype, elucidation of biological and disease pathways as well as experimental aspects of interventions, therapeutics, and precision medicine. The journal aims to publish high quality original papers that present novel findings in all areas of mammalian genetic research as well as review articles on areas of topical interest. The journal will also feature commentaries and editorials to inform readers of breakthrough discoveries as well as issues of research standards, policies and ethics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


