Burkhard Tümmler, Sophia Theres Pallenberg, Anna-Maria Dittrich, Simon Y Graeber, Lutz Naehrlich, Olaf Sommerburg, Marcus A Mall
{"title":"高效CFTR调节剂时代囊性纤维化个体化治疗的进展。","authors":"Burkhard Tümmler, Sophia Theres Pallenberg, Anna-Maria Dittrich, Simon Y Graeber, Lutz Naehrlich, Olaf Sommerburg, Marcus A Mall","doi":"10.1186/s40348-025-00194-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Cystic fibrosis (CF) is a systemic disorder of exocrine glands that is caused by mutations in the CFTR gene.</p><p><strong>Main body: </strong>The basic defect in people with CF (pwCF) leads to impaired epithelial transport of chloride and bicarbonate that can be assessed by CFTR biomarkers, i.e. the β-adrenergic sweat rate and sweat chloride concentration (SCC), chloride conductance of the nasal respiratory epithelium (NPD), urine secretion of bicarbonate, intestinal current measurements (ICM) of chloride secretory responses in rectal biopsies and in bioassays of chloride transport in organoids or cell cultures. CFTR modulators are a novel class of drugs that improve defective posttranslational processing, trafficking and function of mutant CFTR. By April 2025, triple combination therapy with the CFTR potentiator ivacaftor (IVA) and the CFTR correctors elexacaftor (ELX) and tezacaftor (TEZ) has been approved in Europe for the treatment of all pwCF who do not carry two minimal function CFTR mutations. Previous phase 3 and post-approval phase 4 studies in pwCF who harbour one or two alleles of the major mutation F508del consistently reported significant improvements of lung function and anthropometry upon initiation of ELX/TEZ/IVA compared to baseline. Normalization of SCC, NPD and ICM correlated with clinical outcomes on the population level, but the restoration of CFTR function was diverse and not predictive for clinical outcome in the individual patient. Theratyping of non-F508del CF genotypes in patient-derived organoids and cell cultures revealed for most cases clinically meaningful increases of CFTR activity upon exposure to ELX/TEZ/IVA. Likewise, every second CF patient with non-F508del genotypes improved in SCC and clinical outcome upon exposure to ELX/TEZ/IVA indicating that triple CFTR modulator therapy is potentially beneficial for all pwCF who do not carry two minimal function CFTR mutations. This group who is not eligible for CFTR modulators may opt for gene addition therapy in the future, as the first-in-human trial with a recombinant lentiviral vector is underway.</p><p><strong>Future directions: </strong>The upcoming generation of pwCF will probably experience a rather normal life in childhood and adolescence. To classify the upcoming personal signatures of CF disease in the times of efficient modulators, we need more sensitive CFTR biomarkers that address the long-term course of airway and gut microbiome, host defense, epithelial homeostasis and multiorgan metabolism.</p>","PeriodicalId":74215,"journal":{"name":"Molecular and cellular pediatrics","volume":"12 1","pages":"6"},"PeriodicalIF":3.4000,"publicationDate":"2025-05-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12050259/pdf/","citationCount":"0","resultStr":"{\"title\":\"Progress of personalized medicine of cystic fibrosis in the times of efficient CFTR modulators.\",\"authors\":\"Burkhard Tümmler, Sophia Theres Pallenberg, Anna-Maria Dittrich, Simon Y Graeber, Lutz Naehrlich, Olaf Sommerburg, Marcus A Mall\",\"doi\":\"10.1186/s40348-025-00194-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Cystic fibrosis (CF) is a systemic disorder of exocrine glands that is caused by mutations in the CFTR gene.</p><p><strong>Main body: </strong>The basic defect in people with CF (pwCF) leads to impaired epithelial transport of chloride and bicarbonate that can be assessed by CFTR biomarkers, i.e. the β-adrenergic sweat rate and sweat chloride concentration (SCC), chloride conductance of the nasal respiratory epithelium (NPD), urine secretion of bicarbonate, intestinal current measurements (ICM) of chloride secretory responses in rectal biopsies and in bioassays of chloride transport in organoids or cell cultures. CFTR modulators are a novel class of drugs that improve defective posttranslational processing, trafficking and function of mutant CFTR. By April 2025, triple combination therapy with the CFTR potentiator ivacaftor (IVA) and the CFTR correctors elexacaftor (ELX) and tezacaftor (TEZ) has been approved in Europe for the treatment of all pwCF who do not carry two minimal function CFTR mutations. Previous phase 3 and post-approval phase 4 studies in pwCF who harbour one or two alleles of the major mutation F508del consistently reported significant improvements of lung function and anthropometry upon initiation of ELX/TEZ/IVA compared to baseline. Normalization of SCC, NPD and ICM correlated with clinical outcomes on the population level, but the restoration of CFTR function was diverse and not predictive for clinical outcome in the individual patient. Theratyping of non-F508del CF genotypes in patient-derived organoids and cell cultures revealed for most cases clinically meaningful increases of CFTR activity upon exposure to ELX/TEZ/IVA. Likewise, every second CF patient with non-F508del genotypes improved in SCC and clinical outcome upon exposure to ELX/TEZ/IVA indicating that triple CFTR modulator therapy is potentially beneficial for all pwCF who do not carry two minimal function CFTR mutations. This group who is not eligible for CFTR modulators may opt for gene addition therapy in the future, as the first-in-human trial with a recombinant lentiviral vector is underway.</p><p><strong>Future directions: </strong>The upcoming generation of pwCF will probably experience a rather normal life in childhood and adolescence. To classify the upcoming personal signatures of CF disease in the times of efficient modulators, we need more sensitive CFTR biomarkers that address the long-term course of airway and gut microbiome, host defense, epithelial homeostasis and multiorgan metabolism.</p>\",\"PeriodicalId\":74215,\"journal\":{\"name\":\"Molecular and cellular pediatrics\",\"volume\":\"12 1\",\"pages\":\"6\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2025-05-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12050259/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular and cellular pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1186/s40348-025-00194-0\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular and cellular pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s40348-025-00194-0","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
Progress of personalized medicine of cystic fibrosis in the times of efficient CFTR modulators.
Background: Cystic fibrosis (CF) is a systemic disorder of exocrine glands that is caused by mutations in the CFTR gene.
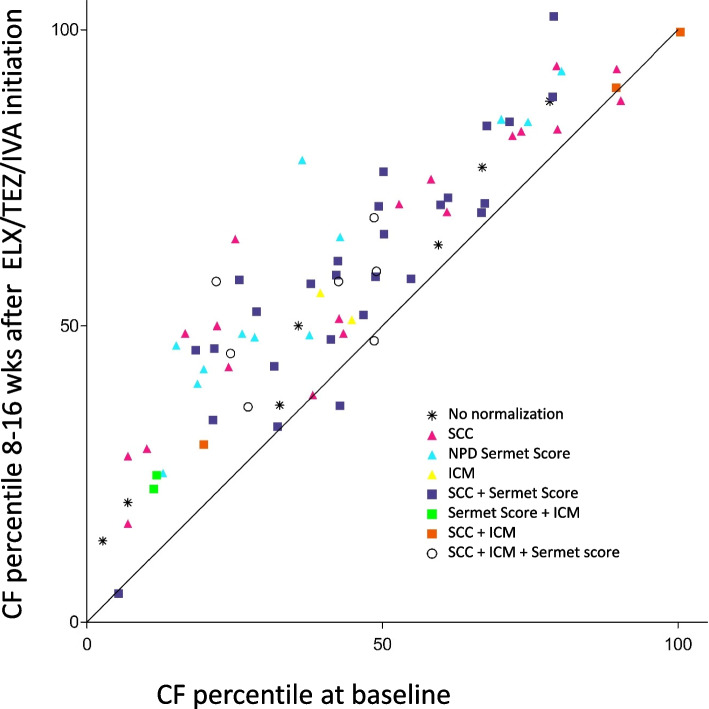
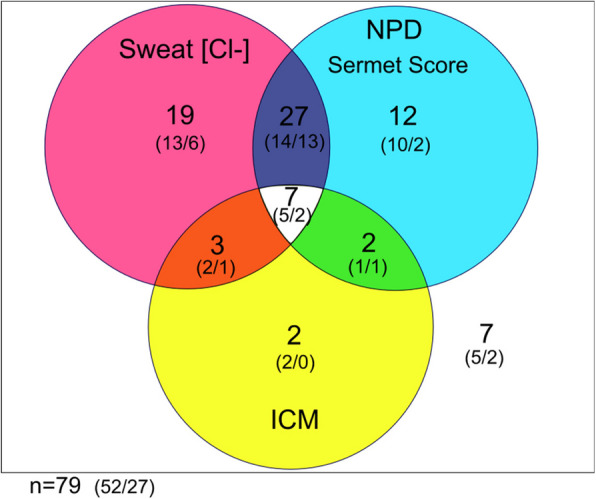
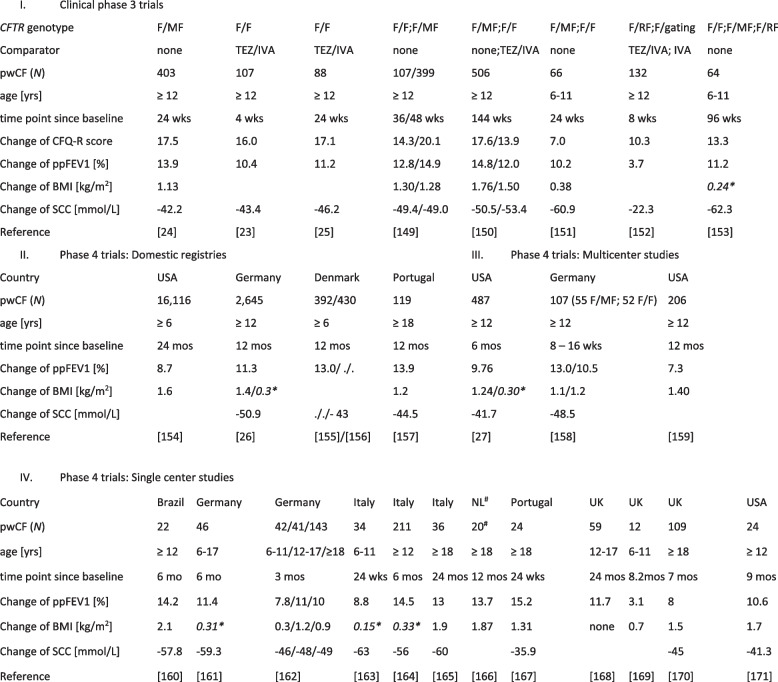
Main body: The basic defect in people with CF (pwCF) leads to impaired epithelial transport of chloride and bicarbonate that can be assessed by CFTR biomarkers, i.e. the β-adrenergic sweat rate and sweat chloride concentration (SCC), chloride conductance of the nasal respiratory epithelium (NPD), urine secretion of bicarbonate, intestinal current measurements (ICM) of chloride secretory responses in rectal biopsies and in bioassays of chloride transport in organoids or cell cultures. CFTR modulators are a novel class of drugs that improve defective posttranslational processing, trafficking and function of mutant CFTR. By April 2025, triple combination therapy with the CFTR potentiator ivacaftor (IVA) and the CFTR correctors elexacaftor (ELX) and tezacaftor (TEZ) has been approved in Europe for the treatment of all pwCF who do not carry two minimal function CFTR mutations. Previous phase 3 and post-approval phase 4 studies in pwCF who harbour one or two alleles of the major mutation F508del consistently reported significant improvements of lung function and anthropometry upon initiation of ELX/TEZ/IVA compared to baseline. Normalization of SCC, NPD and ICM correlated with clinical outcomes on the population level, but the restoration of CFTR function was diverse and not predictive for clinical outcome in the individual patient. Theratyping of non-F508del CF genotypes in patient-derived organoids and cell cultures revealed for most cases clinically meaningful increases of CFTR activity upon exposure to ELX/TEZ/IVA. Likewise, every second CF patient with non-F508del genotypes improved in SCC and clinical outcome upon exposure to ELX/TEZ/IVA indicating that triple CFTR modulator therapy is potentially beneficial for all pwCF who do not carry two minimal function CFTR mutations. This group who is not eligible for CFTR modulators may opt for gene addition therapy in the future, as the first-in-human trial with a recombinant lentiviral vector is underway.
Future directions: The upcoming generation of pwCF will probably experience a rather normal life in childhood and adolescence. To classify the upcoming personal signatures of CF disease in the times of efficient modulators, we need more sensitive CFTR biomarkers that address the long-term course of airway and gut microbiome, host defense, epithelial homeostasis and multiorgan metabolism.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


