Mahsa Saadat, Fatemeh Zare-Mirakabad, Ali Masoudi-Nejad, Mohammad Farahanchi Baradaran, Nazanin Hosseinkhan
{"title":"HLAPepBinder:预测hla -肽结合的集成模型。","authors":"Mahsa Saadat, Fatemeh Zare-Mirakabad, Ali Masoudi-Nejad, Mohammad Farahanchi Baradaran, Nazanin Hosseinkhan","doi":"10.30498/ijb.2024.459448.3927","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Human leukocyte antigens (HLAs) play a pivotal role in orchestrating the host's immune response, offering a promising avenue with reduced adverse effects compared to conventional treatments. Cancer immunotherapies use HLA class I molecules for T cells to recognize tumor antigens, emphasizing the importance of identifying peptides that bind effectively to HLAs. Computer modeling of HLA-peptide binding speeds up the search for immunogenic epitopes, which enhances the prospect of personalized medicine and targeted therapies. The Immune Epitope Database (IEDB) is a vital repository, housing curated immune epitope data and prediction tools for HLA-peptide binding. It can be challenging for immunologists to choose the best tool from the IEDB for predicting HLA-peptide binding. This has led to the creation of consensus-based methods that combine the results of several predictors. One of the major challenges in these methods is how to effectively integrate the results from multiple predictors.</p><p><strong>Objectives: </strong>Previous consensus-based methods integrate at most three tools by relying on simple strategies, such as selecting prediction methods based on their proximity to HLA in training data. In this study, we introduce HLAPepBinder, a novel consensus approach using ensemble machine learning methods to predict HLA-peptide binding, addressing the challenges biologists face in model selection.</p><p><strong>Materials and methods: </strong>The key contribution is the development of an automatic pipeline named <i>HLAPepBinder</i> that integrates the predictions of multiple models using a random forest approach. Unlike previous approaches, <i>HLAPepBinder</i> seamlessly integrates results from all nine predictors, providing a comprehensive and accurate predictive framework. By combining the strengths of these models, <i>HLAPepBinder</i> eliminates the need for manual model selection, providing a streamlined and reliable solution for biologists.</p><p><strong>Results: </strong><i>HLAPepBinder</i> offers a practical and high-performing alternative for HLA-peptide binding predictions, outperforming both traditional methods and complex deep learning models. Compared to the recently introduced transformer-based model, TranspHLA, which requires substantial computational resources, <i>HLAPepBinder</i> demonstrates superior performance in both prediction accuracy and resource efficiency. Notably, it operates effectively in limited computational environments, making it accessible to researchers with minimal resources. The codes are available online at https://github.com/CBRC-lab/HLAPepBinder.</p><p><strong>Conclusion: </strong>Our study introduces a novel ensemble-learning model designed to enhance the accuracy and efficiency of HLA-peptide binding predictions. Due to the lack of reliable negative data and the typical assumption of unknown interactions being negative, we focus on analyzing the unknown HLA-peptide bindings in the test set that our model predicts with 100% certainty as positive bindings. Using <i>HLAPepBinder</i>, we identify 26 HLA-peptide pairs with absolute prediction confidence. These predictions are validated through a multi-step pipeline involving literature review, BLAST sequence similarity analysis, and molecular docking studies. This comprehensive validation process highlights <i>HLAPepBinder</i>'s ability to make accurate and reliable predictions, contributing significantly to advancements in immunotherapy and vaccine development.</p>","PeriodicalId":14492,"journal":{"name":"Iranian Journal of Biotechnology","volume":"22 4","pages":"e3927"},"PeriodicalIF":1.5000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11993240/pdf/","citationCount":"0","resultStr":"{\"title\":\"<i>HLAPepBinder</i>: An Ensemble Model for The Prediction Of HLA-Peptide Binding.\",\"authors\":\"Mahsa Saadat, Fatemeh Zare-Mirakabad, Ali Masoudi-Nejad, Mohammad Farahanchi Baradaran, Nazanin Hosseinkhan\",\"doi\":\"10.30498/ijb.2024.459448.3927\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Human leukocyte antigens (HLAs) play a pivotal role in orchestrating the host's immune response, offering a promising avenue with reduced adverse effects compared to conventional treatments. Cancer immunotherapies use HLA class I molecules for T cells to recognize tumor antigens, emphasizing the importance of identifying peptides that bind effectively to HLAs. Computer modeling of HLA-peptide binding speeds up the search for immunogenic epitopes, which enhances the prospect of personalized medicine and targeted therapies. The Immune Epitope Database (IEDB) is a vital repository, housing curated immune epitope data and prediction tools for HLA-peptide binding. It can be challenging for immunologists to choose the best tool from the IEDB for predicting HLA-peptide binding. This has led to the creation of consensus-based methods that combine the results of several predictors. One of the major challenges in these methods is how to effectively integrate the results from multiple predictors.</p><p><strong>Objectives: </strong>Previous consensus-based methods integrate at most three tools by relying on simple strategies, such as selecting prediction methods based on their proximity to HLA in training data. In this study, we introduce HLAPepBinder, a novel consensus approach using ensemble machine learning methods to predict HLA-peptide binding, addressing the challenges biologists face in model selection.</p><p><strong>Materials and methods: </strong>The key contribution is the development of an automatic pipeline named <i>HLAPepBinder</i> that integrates the predictions of multiple models using a random forest approach. Unlike previous approaches, <i>HLAPepBinder</i> seamlessly integrates results from all nine predictors, providing a comprehensive and accurate predictive framework. By combining the strengths of these models, <i>HLAPepBinder</i> eliminates the need for manual model selection, providing a streamlined and reliable solution for biologists.</p><p><strong>Results: </strong><i>HLAPepBinder</i> offers a practical and high-performing alternative for HLA-peptide binding predictions, outperforming both traditional methods and complex deep learning models. Compared to the recently introduced transformer-based model, TranspHLA, which requires substantial computational resources, <i>HLAPepBinder</i> demonstrates superior performance in both prediction accuracy and resource efficiency. Notably, it operates effectively in limited computational environments, making it accessible to researchers with minimal resources. The codes are available online at https://github.com/CBRC-lab/HLAPepBinder.</p><p><strong>Conclusion: </strong>Our study introduces a novel ensemble-learning model designed to enhance the accuracy and efficiency of HLA-peptide binding predictions. Due to the lack of reliable negative data and the typical assumption of unknown interactions being negative, we focus on analyzing the unknown HLA-peptide bindings in the test set that our model predicts with 100% certainty as positive bindings. Using <i>HLAPepBinder</i>, we identify 26 HLA-peptide pairs with absolute prediction confidence. These predictions are validated through a multi-step pipeline involving literature review, BLAST sequence similarity analysis, and molecular docking studies. This comprehensive validation process highlights <i>HLAPepBinder</i>'s ability to make accurate and reliable predictions, contributing significantly to advancements in immunotherapy and vaccine development.</p>\",\"PeriodicalId\":14492,\"journal\":{\"name\":\"Iranian Journal of Biotechnology\",\"volume\":\"22 4\",\"pages\":\"e3927\"},\"PeriodicalIF\":1.5000,\"publicationDate\":\"2024-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11993240/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Iranian Journal of Biotechnology\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://doi.org/10.30498/ijb.2024.459448.3927\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOTECHNOLOGY & APPLIED MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Iranian Journal of Biotechnology","FirstCategoryId":"5","ListUrlMain":"https://doi.org/10.30498/ijb.2024.459448.3927","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOTECHNOLOGY & APPLIED MICROBIOLOGY","Score":null,"Total":0}
HLAPepBinder: An Ensemble Model for The Prediction Of HLA-Peptide Binding.
Background: Human leukocyte antigens (HLAs) play a pivotal role in orchestrating the host's immune response, offering a promising avenue with reduced adverse effects compared to conventional treatments. Cancer immunotherapies use HLA class I molecules for T cells to recognize tumor antigens, emphasizing the importance of identifying peptides that bind effectively to HLAs. Computer modeling of HLA-peptide binding speeds up the search for immunogenic epitopes, which enhances the prospect of personalized medicine and targeted therapies. The Immune Epitope Database (IEDB) is a vital repository, housing curated immune epitope data and prediction tools for HLA-peptide binding. It can be challenging for immunologists to choose the best tool from the IEDB for predicting HLA-peptide binding. This has led to the creation of consensus-based methods that combine the results of several predictors. One of the major challenges in these methods is how to effectively integrate the results from multiple predictors.
Objectives: Previous consensus-based methods integrate at most three tools by relying on simple strategies, such as selecting prediction methods based on their proximity to HLA in training data. In this study, we introduce HLAPepBinder, a novel consensus approach using ensemble machine learning methods to predict HLA-peptide binding, addressing the challenges biologists face in model selection.
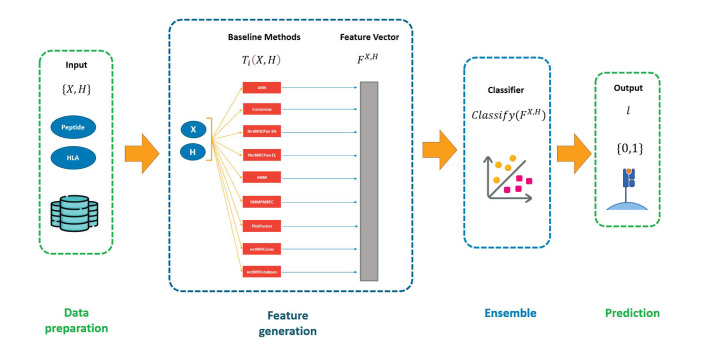
Materials and methods: The key contribution is the development of an automatic pipeline named HLAPepBinder that integrates the predictions of multiple models using a random forest approach. Unlike previous approaches, HLAPepBinder seamlessly integrates results from all nine predictors, providing a comprehensive and accurate predictive framework. By combining the strengths of these models, HLAPepBinder eliminates the need for manual model selection, providing a streamlined and reliable solution for biologists.
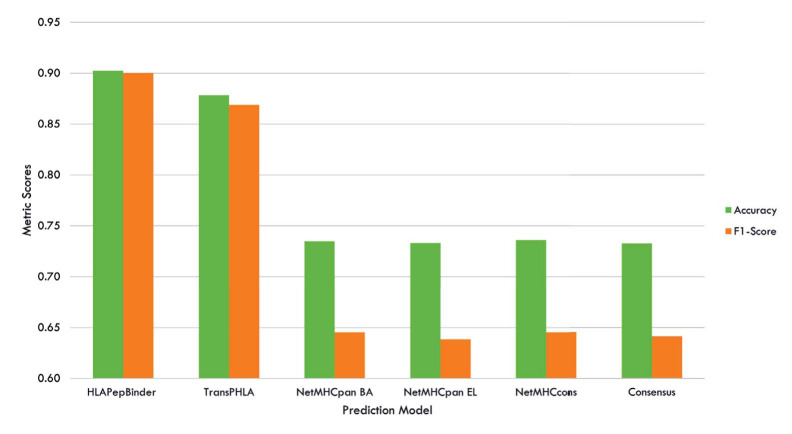
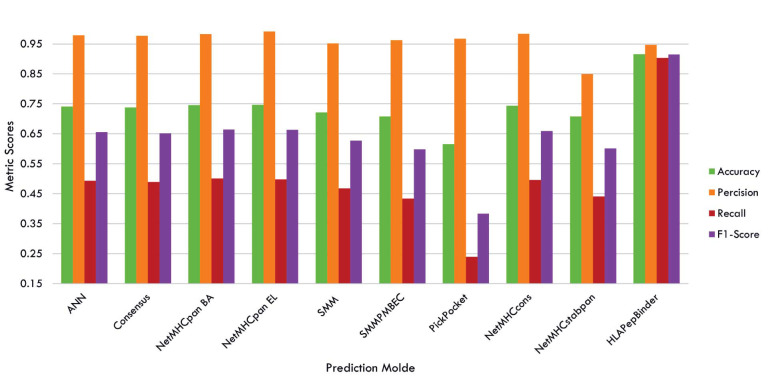
Results: HLAPepBinder offers a practical and high-performing alternative for HLA-peptide binding predictions, outperforming both traditional methods and complex deep learning models. Compared to the recently introduced transformer-based model, TranspHLA, which requires substantial computational resources, HLAPepBinder demonstrates superior performance in both prediction accuracy and resource efficiency. Notably, it operates effectively in limited computational environments, making it accessible to researchers with minimal resources. The codes are available online at https://github.com/CBRC-lab/HLAPepBinder.
Conclusion: Our study introduces a novel ensemble-learning model designed to enhance the accuracy and efficiency of HLA-peptide binding predictions. Due to the lack of reliable negative data and the typical assumption of unknown interactions being negative, we focus on analyzing the unknown HLA-peptide bindings in the test set that our model predicts with 100% certainty as positive bindings. Using HLAPepBinder, we identify 26 HLA-peptide pairs with absolute prediction confidence. These predictions are validated through a multi-step pipeline involving literature review, BLAST sequence similarity analysis, and molecular docking studies. This comprehensive validation process highlights HLAPepBinder's ability to make accurate and reliable predictions, contributing significantly to advancements in immunotherapy and vaccine development.
期刊介绍:
Iranian Journal of Biotechnology (IJB) is published quarterly by the National Institute of Genetic Engineering and Biotechnology. IJB publishes original scientific research papers in the broad area of Biotechnology such as, Agriculture, Animal and Marine Sciences, Basic Sciences, Bioinformatics, Biosafety and Bioethics, Environment, Industry and Mining and Medical Sciences.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


