{"title":"以地高辛、非索非那定和达比加群为探测药物的p -糖蛋白抑制剂和诱导剂在人体中的作用的系统评价和分类。","authors":"Claire Coumau, Chantal Csajka","doi":"10.1007/s40262-025-01514-3","DOIUrl":null,"url":null,"abstract":"<p><p>P-glycoprotein is a critical efflux transporter that may significantly affect the pharmacokinetics of various drugs by influencing their absorption, distribution and elimination. While European and American regulatory guidelines provide lists of P-glycoprotein modulators, they lack specificity concerning in vivo studies and clear guidance on inducers, creating uncertainty in their clinical relevance. A systematic search on in vivo clinical studies involving healthy volunteers using fexofenadine, dabigatran and digoxin as P-glycoprotein substrates has been performed in accordance with the PRISMA guidelines. A total of 151 studies assessing the impact of P-glycoprotein modulators on the concentration-time profile of P-glycoprotein substrates were retrieved. Additionally, data on the P-glycoprotein modulators' effect on cytochrome P450 3A4 induction or inhibition were also collected. P-gp modulators were classified as potent, moderate, weak or non-interactors for P-glycoprotein, with or without cytochrome P450 3A4 impact, on the basis of the area under the concentration-time curve ratio. This classification was adapted from the Food and Drug Administration criteria for cytochrome interactions. This systematic review identified 49 area under the plasma concentration-time curve ratio values corresponding to P-glycoprotein inhibitors, 23 to P-glycoprotein inducers and 131 to non-interactors. Of these, only 32.5% and 41.1% were classified as weak to potent, respectively. Only 0.7% of inhibitors and no inducers were classified as potent. This suggests that most P-glycoprotein modulators have a limited impact on drug exposure. The potential for interaction increases when P-glycoprotein modulators also affect cytochrome P450 3A4, which is the case for 59.9% of P-glycoprotein modulators. However, some moderate P-glycoprotein modulators may have clinically significant effects depending on the therapeutic margin of the substrate and the clinical context.</p>","PeriodicalId":10405,"journal":{"name":"Clinical Pharmacokinetics","volume":" ","pages":"849-863"},"PeriodicalIF":4.0000,"publicationDate":"2025-06-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12158833/pdf/","citationCount":"0","resultStr":"{\"title\":\"A Systematic Review and Classification of the Effects of P-glycoprotein Inhibitors and Inducers in Humans, Using Digoxin, Fexofenadine, and Dabigatran as Probe Drugs.\",\"authors\":\"Claire Coumau, Chantal Csajka\",\"doi\":\"10.1007/s40262-025-01514-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>P-glycoprotein is a critical efflux transporter that may significantly affect the pharmacokinetics of various drugs by influencing their absorption, distribution and elimination. While European and American regulatory guidelines provide lists of P-glycoprotein modulators, they lack specificity concerning in vivo studies and clear guidance on inducers, creating uncertainty in their clinical relevance. A systematic search on in vivo clinical studies involving healthy volunteers using fexofenadine, dabigatran and digoxin as P-glycoprotein substrates has been performed in accordance with the PRISMA guidelines. A total of 151 studies assessing the impact of P-glycoprotein modulators on the concentration-time profile of P-glycoprotein substrates were retrieved. Additionally, data on the P-glycoprotein modulators' effect on cytochrome P450 3A4 induction or inhibition were also collected. P-gp modulators were classified as potent, moderate, weak or non-interactors for P-glycoprotein, with or without cytochrome P450 3A4 impact, on the basis of the area under the concentration-time curve ratio. This classification was adapted from the Food and Drug Administration criteria for cytochrome interactions. This systematic review identified 49 area under the plasma concentration-time curve ratio values corresponding to P-glycoprotein inhibitors, 23 to P-glycoprotein inducers and 131 to non-interactors. Of these, only 32.5% and 41.1% were classified as weak to potent, respectively. Only 0.7% of inhibitors and no inducers were classified as potent. This suggests that most P-glycoprotein modulators have a limited impact on drug exposure. The potential for interaction increases when P-glycoprotein modulators also affect cytochrome P450 3A4, which is the case for 59.9% of P-glycoprotein modulators. However, some moderate P-glycoprotein modulators may have clinically significant effects depending on the therapeutic margin of the substrate and the clinical context.</p>\",\"PeriodicalId\":10405,\"journal\":{\"name\":\"Clinical Pharmacokinetics\",\"volume\":\" \",\"pages\":\"849-863\"},\"PeriodicalIF\":4.0000,\"publicationDate\":\"2025-06-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12158833/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pharmacokinetics\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s40262-025-01514-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/5/11 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PHARMACOLOGY & PHARMACY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pharmacokinetics","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s40262-025-01514-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/11 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
A Systematic Review and Classification of the Effects of P-glycoprotein Inhibitors and Inducers in Humans, Using Digoxin, Fexofenadine, and Dabigatran as Probe Drugs.
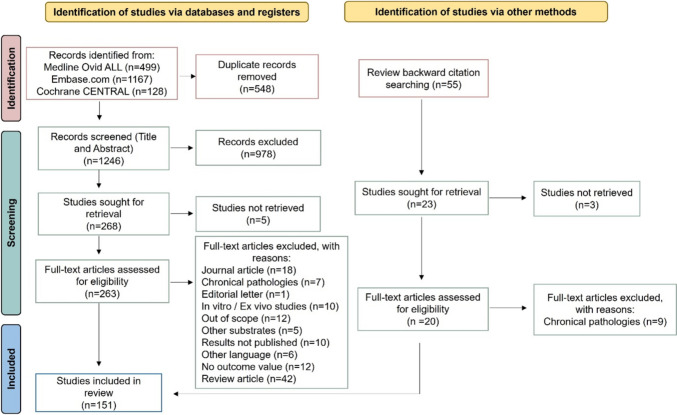
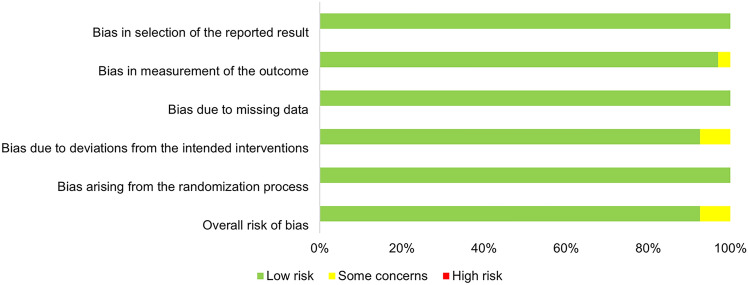
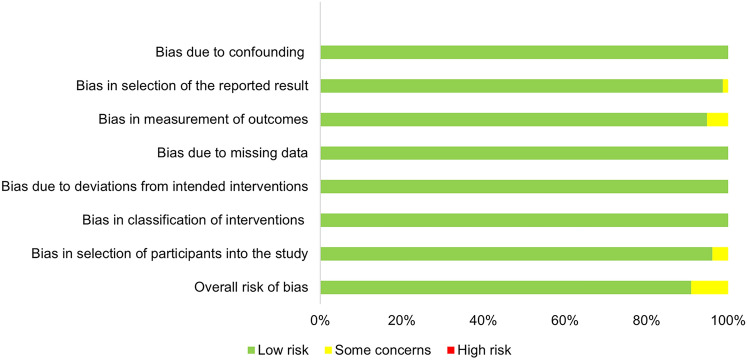
P-glycoprotein is a critical efflux transporter that may significantly affect the pharmacokinetics of various drugs by influencing their absorption, distribution and elimination. While European and American regulatory guidelines provide lists of P-glycoprotein modulators, they lack specificity concerning in vivo studies and clear guidance on inducers, creating uncertainty in their clinical relevance. A systematic search on in vivo clinical studies involving healthy volunteers using fexofenadine, dabigatran and digoxin as P-glycoprotein substrates has been performed in accordance with the PRISMA guidelines. A total of 151 studies assessing the impact of P-glycoprotein modulators on the concentration-time profile of P-glycoprotein substrates were retrieved. Additionally, data on the P-glycoprotein modulators' effect on cytochrome P450 3A4 induction or inhibition were also collected. P-gp modulators were classified as potent, moderate, weak or non-interactors for P-glycoprotein, with or without cytochrome P450 3A4 impact, on the basis of the area under the concentration-time curve ratio. This classification was adapted from the Food and Drug Administration criteria for cytochrome interactions. This systematic review identified 49 area under the plasma concentration-time curve ratio values corresponding to P-glycoprotein inhibitors, 23 to P-glycoprotein inducers and 131 to non-interactors. Of these, only 32.5% and 41.1% were classified as weak to potent, respectively. Only 0.7% of inhibitors and no inducers were classified as potent. This suggests that most P-glycoprotein modulators have a limited impact on drug exposure. The potential for interaction increases when P-glycoprotein modulators also affect cytochrome P450 3A4, which is the case for 59.9% of P-glycoprotein modulators. However, some moderate P-glycoprotein modulators may have clinically significant effects depending on the therapeutic margin of the substrate and the clinical context.
期刊介绍:
Clinical Pharmacokinetics promotes the continuing development of clinical pharmacokinetics and pharmacodynamics for the improvement of drug therapy, and for furthering postgraduate education in clinical pharmacology and therapeutics.
Pharmacokinetics, the study of drug disposition in the body, is an integral part of drug development and rational use. Knowledge and application of pharmacokinetic principles leads to accelerated drug development, cost effective drug use and a reduced frequency of adverse effects and drug interactions.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


