{"title":"使用霉酚酸酯和利妥昔单抗维持治疗的重叠综合征硬肌炎患者1年以上完全恢复,停药8个月内复发。","authors":"Inês Amarante, Catarina Faustino, Bruna Mota, Mariana Cascais, Jorge Almeida, Marta Valentim","doi":"10.12890/2025_005154","DOIUrl":null,"url":null,"abstract":"<p><p>Scleromyositis (SM) is an emerging, distinct entity within the spectrum of diffuse systemic sclerosis (SSc) and autoimmune inflammatory myopathies. It can carry a poorer prognosis due to multisystem involvement and extramuscular complications, with no consensus on treatment strategies currently available. We report on a 20-year-old woman with a history of recurrent infections over the past year, who presented with persistent myalgias for four months. On examination, she exhibited muscle weakness in the scapular and pelvic girdles, along with sclerodactyly. Laboratory results showed elevated creatine kinase (13,000 U/l), aldolase (15.6 U/l), lactate dehydrogenase (1,481 U/l) and myoglobin (4,400 ng/ml). Autoimmune screening was positive for antinuclear antibodies, anti-PM-scleroderma (anti-PM-Scl) 75 and 100, and anti-CENP-B antibodies. An MRI of the pelvic girdle indicated acute/subacute myositis, and electromyography revealed both distal and proximal myopathy. Muscle biopsy showed extensive necrosis with minimal inflammatory infiltration. Nailfold capillaroscopy demonstrated an early scleroderma pattern, while CT and spirometry revealed mild interstitial restrictive lung disease. Initial treatment involved intravenous immunoglobulin (IVIG), mycophenolate mofetil (MMF) and prednisolone. This was followed by maintenance therapy with MMF and rituximab (RTX) every six months for the first year. Over six months, the patient showed progressive improvement in muscle strength and normalisation of muscle enzyme levels. In conclusion, SM presents with variable phenotypes, ranging from mild to extensive systemic involvement. This case underscores the importance of individualised patient stratification and highlights the need for structured induction and maintenance therapy due to the disease's extensive activity.</p><p><strong>Learning points: </strong>Scleromyositis (SM) is a rare and poorly understood condition that combines features of systemic sclerosis and immune-mediated myositis, often linked to anti-PM-scleroderma antibodies.Despite efforts to classify SM, it demonstrates unique clinical patterns that distinguish it from other myositis subtypes.The management of SM remains challenging, as there are no established guidelines or standardised treatment protocols.</p>","PeriodicalId":11908,"journal":{"name":"European journal of case reports in internal medicine","volume":"12 5","pages":"005154"},"PeriodicalIF":0.0000,"publicationDate":"2025-05-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12061208/pdf/","citationCount":"0","resultStr":"{\"title\":\"Full Recovery Over One Year of the Overlap Syndrome Scleromyositis to Maintenance Therapy with Mycophenolate Mofetil and Rituximab, which Relapsed within 8 Months of Discontinuation.\",\"authors\":\"Inês Amarante, Catarina Faustino, Bruna Mota, Mariana Cascais, Jorge Almeida, Marta Valentim\",\"doi\":\"10.12890/2025_005154\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Scleromyositis (SM) is an emerging, distinct entity within the spectrum of diffuse systemic sclerosis (SSc) and autoimmune inflammatory myopathies. It can carry a poorer prognosis due to multisystem involvement and extramuscular complications, with no consensus on treatment strategies currently available. We report on a 20-year-old woman with a history of recurrent infections over the past year, who presented with persistent myalgias for four months. On examination, she exhibited muscle weakness in the scapular and pelvic girdles, along with sclerodactyly. Laboratory results showed elevated creatine kinase (13,000 U/l), aldolase (15.6 U/l), lactate dehydrogenase (1,481 U/l) and myoglobin (4,400 ng/ml). Autoimmune screening was positive for antinuclear antibodies, anti-PM-scleroderma (anti-PM-Scl) 75 and 100, and anti-CENP-B antibodies. An MRI of the pelvic girdle indicated acute/subacute myositis, and electromyography revealed both distal and proximal myopathy. Muscle biopsy showed extensive necrosis with minimal inflammatory infiltration. Nailfold capillaroscopy demonstrated an early scleroderma pattern, while CT and spirometry revealed mild interstitial restrictive lung disease. Initial treatment involved intravenous immunoglobulin (IVIG), mycophenolate mofetil (MMF) and prednisolone. This was followed by maintenance therapy with MMF and rituximab (RTX) every six months for the first year. Over six months, the patient showed progressive improvement in muscle strength and normalisation of muscle enzyme levels. In conclusion, SM presents with variable phenotypes, ranging from mild to extensive systemic involvement. This case underscores the importance of individualised patient stratification and highlights the need for structured induction and maintenance therapy due to the disease's extensive activity.</p><p><strong>Learning points: </strong>Scleromyositis (SM) is a rare and poorly understood condition that combines features of systemic sclerosis and immune-mediated myositis, often linked to anti-PM-scleroderma antibodies.Despite efforts to classify SM, it demonstrates unique clinical patterns that distinguish it from other myositis subtypes.The management of SM remains challenging, as there are no established guidelines or standardised treatment protocols.</p>\",\"PeriodicalId\":11908,\"journal\":{\"name\":\"European journal of case reports in internal medicine\",\"volume\":\"12 5\",\"pages\":\"005154\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-05-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12061208/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"European journal of case reports in internal medicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.12890/2025_005154\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"Medicine\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"European journal of case reports in internal medicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.12890/2025_005154","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"Medicine","Score":null,"Total":0}
Full Recovery Over One Year of the Overlap Syndrome Scleromyositis to Maintenance Therapy with Mycophenolate Mofetil and Rituximab, which Relapsed within 8 Months of Discontinuation.
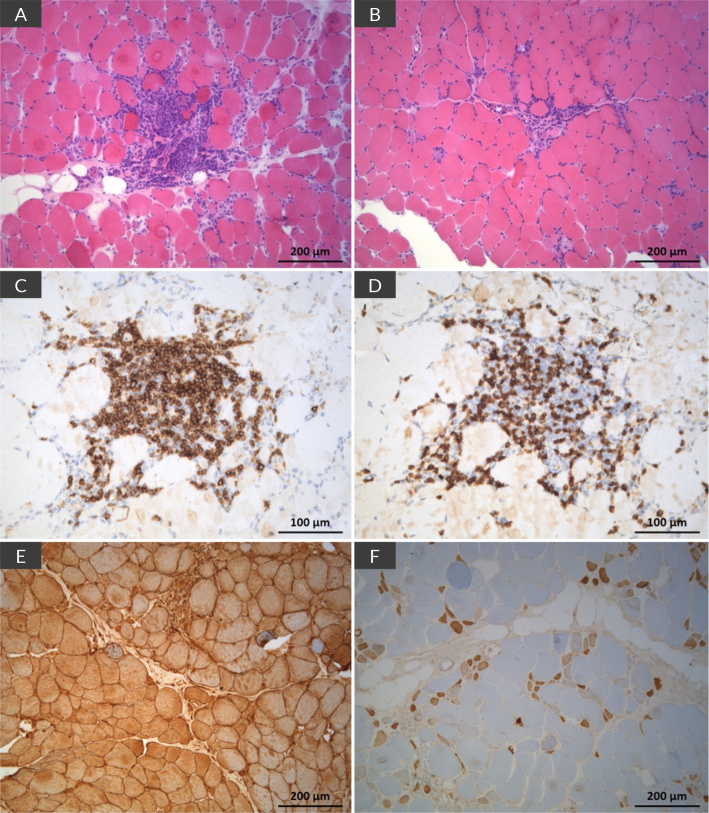
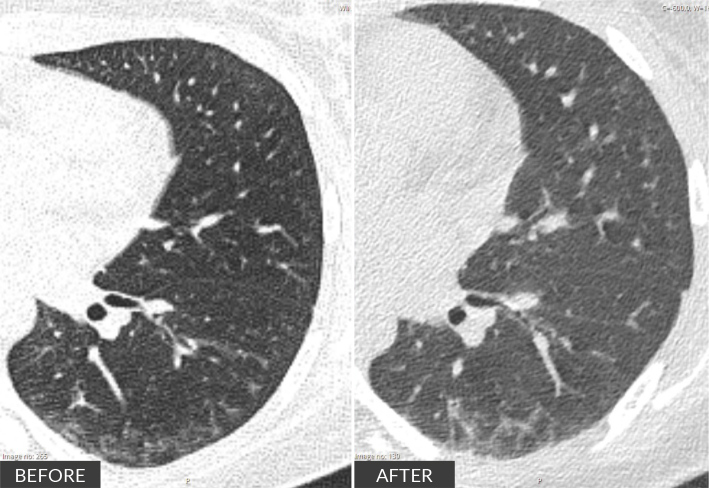
Scleromyositis (SM) is an emerging, distinct entity within the spectrum of diffuse systemic sclerosis (SSc) and autoimmune inflammatory myopathies. It can carry a poorer prognosis due to multisystem involvement and extramuscular complications, with no consensus on treatment strategies currently available. We report on a 20-year-old woman with a history of recurrent infections over the past year, who presented with persistent myalgias for four months. On examination, she exhibited muscle weakness in the scapular and pelvic girdles, along with sclerodactyly. Laboratory results showed elevated creatine kinase (13,000 U/l), aldolase (15.6 U/l), lactate dehydrogenase (1,481 U/l) and myoglobin (4,400 ng/ml). Autoimmune screening was positive for antinuclear antibodies, anti-PM-scleroderma (anti-PM-Scl) 75 and 100, and anti-CENP-B antibodies. An MRI of the pelvic girdle indicated acute/subacute myositis, and electromyography revealed both distal and proximal myopathy. Muscle biopsy showed extensive necrosis with minimal inflammatory infiltration. Nailfold capillaroscopy demonstrated an early scleroderma pattern, while CT and spirometry revealed mild interstitial restrictive lung disease. Initial treatment involved intravenous immunoglobulin (IVIG), mycophenolate mofetil (MMF) and prednisolone. This was followed by maintenance therapy with MMF and rituximab (RTX) every six months for the first year. Over six months, the patient showed progressive improvement in muscle strength and normalisation of muscle enzyme levels. In conclusion, SM presents with variable phenotypes, ranging from mild to extensive systemic involvement. This case underscores the importance of individualised patient stratification and highlights the need for structured induction and maintenance therapy due to the disease's extensive activity.
Learning points: Scleromyositis (SM) is a rare and poorly understood condition that combines features of systemic sclerosis and immune-mediated myositis, often linked to anti-PM-scleroderma antibodies.Despite efforts to classify SM, it demonstrates unique clinical patterns that distinguish it from other myositis subtypes.The management of SM remains challenging, as there are no established guidelines or standardised treatment protocols.
期刊介绍:
The European Journal of Case Reports in Internal Medicine is an official journal of the European Federation of Internal Medicine (EFIM), representing 35 national societies from 33 European countries. The Journal''s mission is to promote the best medical practice and innovation in the field of acute and general medicine. It also provides a forum for internal medicine doctors where they can share new approaches with the aim of improving diagnostic and clinical skills in this field. EJCRIM welcomes high-quality case reports describing unusual or complex cases that an internist may encounter in everyday practice. The cases should either demonstrate the appropriateness of a diagnostic/therapeutic approach, describe a new procedure or maneuver, or show unusual manifestations of a disease or unexpected reactions. The Journal only accepts and publishes those case reports whose learning points provide new insight and/or contribute to advancing medical knowledge both in terms of diagnostics and therapeutic approaches. Case reports of medical errors, therefore, are also welcome as long as they provide innovative measures on how to prevent them in the current practice (Instructive Errors). The Journal may also consider brief and reasoned reports on issues relevant to the practice of Internal Medicine, as well as Abstracts submitted to the scientific meetings of acknowledged medical societies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


