E. Diral, C. Campochiaro, G. Furnari, F. Moretti, J. Ferrari, C. Elena, G. Battipaglia, S. Barbato, A. Vitale, M. Frigeni, F. Crisafulli, M. Frassi, C. Papayannidis, A. D. Romagnoli, C. Cattaneo, A. D'Ambrosio, E. Morsia, P. Musto, G. Rivoli, M. E. Dragani, C. Toffalori, G. M. Bergonzi, G. Scorpio, A. Tomelleri, M. T. Voso, C. Gurnari, L. Vago, L. Dagna, F. Ciceri
{"title":"促红细胞剂治疗VEXAS综合征:一项意大利多中心队列回顾性研究。","authors":"E. Diral, C. Campochiaro, G. Furnari, F. Moretti, J. Ferrari, C. Elena, G. Battipaglia, S. Barbato, A. Vitale, M. Frigeni, F. Crisafulli, M. Frassi, C. Papayannidis, A. D. Romagnoli, C. Cattaneo, A. D'Ambrosio, E. Morsia, P. Musto, G. Rivoli, M. E. Dragani, C. Toffalori, G. M. Bergonzi, G. Scorpio, A. Tomelleri, M. T. Voso, C. Gurnari, L. Vago, L. Dagna, F. Ciceri","doi":"10.1111/bjh.20157","DOIUrl":null,"url":null,"abstract":"<p>VEXAS (Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic) syndrome is an autoinflammatory disorder caused by somatic mutations in the <i>UBA1</i> gene, resulting in impaired protein ubiquitylation.<span><sup>1</sup></span> Patients present with inflammatory symptoms and a variety of haematological conditions, including plasma cell dyscrasias and cytopenias,<span><sup>2</sup></span> mostly macrocytic anaemia.<span><sup>1</sup></span> Accordingly, patients can be categorized as per the WHO 2022 criteria into ICUS (Idiopathic Cytopenia of Uncertain Significance), CCUS (Clonal Cytopenia of Uncertain Significance) or MDS (Myelodysplastic Neoplasia) in up to 50% of patients.<span><sup>3</sup></span></p><p>Currently, no standard therapy is available for VEXAS. For patients with moderate-to-severe anaemia, transfusions and erythropoiesis-stimulating agents (ESAs) are used, borrowing practices from lower risk MDS with the goal of improving quality of life. In this retrospective study, we analyse the use of ESAs in VEXAS patients, their efficacy and safety.</p><p>This study was conducted through a national survey sent to centres known to be involved in the clinical management of VEXAS patients. An electronic questionnaire was distributed from September to December 2024. We included patients with a confirmed diagnosis of VEXAS by <i>UBA1</i> testing receiving ESA treatment. Centres were queried for relevant data concerning clinical manifestations of VEXAS, haematological conditions as per the WHO 2022<span><sup>4</sup></span> and ESA treatment. Response to ESAs was evaluated after at least 16 weeks according to the IWG 2018 criteria.<span><sup>5</sup></span> MDS risk assessment was assigned as per Revised International Prognostic Scoring System (IPSS-R) and its molecular (IPSS-M) version. <i>UBA1</i> variant allele frequency (VAF) was determined by digital droplet PCR (ddPCR).<span><sup>6</sup></span> The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.</p><p>Patients' features were summarized using descriptive statistics. Quantitative variables were expressed as medians and interquartile range (IQR). Qualitative variables were expressed as numbers and percentages. Univariable and multivariable logistic regression analyses with odds ratio (OR) and 95% confidence interval (CI) were applied to analyse the ability of baseline features to predict response to ESA. Analysed variables included: underlying haematological condition; IPSS-R and IPSS-M risk categories; endogenous EPO levels; baseline haemoglobin levels; transfusion dependence; UBA1 clone size; and baseline CRP levels. Analyses, graphs and data visualizations were generated using Microsoft Excel and Graphpad Prism softwares.</p><p>Overall, 32 male patients received ESA treatment in 13 centres. Median age at VEXAS diagnosis was 66 years (IQR: 64–73). Clinical manifestations are reported in Table 1. Regarding <i>UBA1</i> variants, all patients harboured the canonical p.Met41Thr (<i>n</i> = 16, 50%), p.Met41Val (<i>n</i> = 12, 38%) and p.Met41Leu (<i>n</i> = 4, 12%) mutations. Next-generation sequencing (NGS) before ESA initiation was available for 25 patients (78%) and detected additional somatic myeloid mutations in 72% of cases (median of 1 mutation per patient, IQR: 0–1). The most frequently mutated genes belonged to the DTA triad<span><sup>7</sup></span>: DNMT3A, TET2, ASXL1 found in 72% (<i>n</i> = 13), 17% (<i>n</i> = 3) and 22% (<i>n</i> = 4) of cases respectively (Figure 1A). Twenty-six patients (82%) had concomitant MDS (Figure 1B), predominantly MDS-LB according to the WHO 2022 (73%, <i>n</i> = 19, Figure 1C). ICUS and CCUS were diagnosed in 3 (9%) patients each. The majority of MDS belonged to lower risk categories of IPSS-R/M scoring systems (IPSS-R <i>n</i> = 22, 85%; IPSS-M <i>n</i> = 17, 65%; Figure 1D).</p><p>Twenty-eight patients (88%) had symptomatic anaemia at baseline, with median haemoglobin levels of 91 g/L (range: 74–10). Of them, 20 (63%) were transfusion-dependent: 13 had a low transfusion burden (LTB, 41.9%) and 7 (35%) a high transfusion burden (HTB) (Figure 1E).</p><p>Before ESA initiation, <i>UBA1</i> VAF determination by ddPCR was available in 17 patients (53%, median 75%, IQR: 57.84–86.95), median C-reactive protein (CRP) levels were 15.8 mg/L (IQR: 7.6–53) and endogenous erythropoietin (EPO) levels were 124 mU/mL (IQR: 57.4–201). Concomitant treatments are shown in Figure 1F: 97% (<i>N</i> = 30) of patients were receiving VEXAS-directed anti-inflammatory therapy. Azacitidine was administered concomitantly with ESAs in two cases: Both achieved haematological improvement–erythroid (HI-E) and transfusion independence after azacitidine initiation. In these two cases, haemoglobin improvement likely reflects the effect of the hypomethylating agent, though UBA1 clone monitoring data are lacking.</p><p>Overall, 20 (63%) and 9 (28%) patients received epoetin alpha or epoetin zeta, at a median starting dose of 40 000 IU weekly (IQR: 30 000–40 000). Three patients received darbepoetin. In 11 patients (34%), the ESA dose was increased to the maximum of 80 000 IU weekly. Five of 11 patients (45%) receiving the maximum recommended dose eventually achieved a response. The median duration of exposure to ESAs was 41.3 weeks (range: 0.3–205.7) in our cohort.</p><p>HI-E was achieved in 19 patients (59%), with all responders obtaining transfusion independence (Figure S1). Responding patients were mostly LTB (58%, <i>n</i> = 11) or non-transfusion dependent (NTD, 37%, <i>n</i> = 7) and had lower median endogenous EPO levels (123 mU/mL, IQR: 51.2–177.7) compared to patients who had no response (NR; 124 mU/mL, IQR: 67–200), consistent with the Hellström-Lindberg score.<span><sup>8</sup></span> Of the seven HTB patients, only 1 (14%) obtained a major HI-E response, lasting 27 months. Six of 13 non-responding patients (46%) received ESAs full dose (80 000 IU/week) and the duration of exposure to ESA in these patients was >8 weeks. In non-responders, median CRP level changed from 14.5 mg/L prior to therapy (IQR: 9.9–55.9) to 21.3 mg/L at discontinuation (IQR: 10.78–54.4). In patients who lost response, median CRP level was 19.3 mg/L at baseline (IQR: 3–161.95) compared to 19.5 mg/L at ESA discontinuation.</p><p>At multivariate analysis, endogenous EPO was the only variable associated with the probability of response to ESAs (OR: 0.985; 95% CI = 0.972–0.999; <i>p</i> = 0.033); the type of <i>UBA1</i> mutation, as well as concurrent somatic myeloid mutations, did not correlate with response.</p><p>Median duration of response to ESAs was 13 months (IQR: 7.5–26.5). Six patients who initially achieved HI-E lost response (LOR, 31%) after a median of 14 months (IQR: 9.5–27.75). Median follow-up for responding patients was 15.6 months (IQR: 7.9–27.7 months).</p><p>Overall, 15/19 patients (79%) were transfusion dependent at ESA discontinuation due to NR or LOR. ddPCR for UBA1 was available at ESA discontinuation for four patients, and no significant changes were observed (Figure 1G).</p><p>For patients who had NR or LOR, subsequent treatments after ESA discontinuation included biological disease-modifying anti-rheumatic drugs (bDMARDs, <i>n</i> = 6, 31%), azacitidine alone or in combination with bDMARDSs (<i>n</i> = 4, 21%), allogeneic haematopoietic cell transplantation (allo-HCT, <i>n</i> = 6, 31%) or supportive care (<i>n</i> = 2, 10%), including red blood cell transfusions. Patients with long-lasting response to ESAs also underwent allo-HCT (<i>n</i> = 1) or azacitidine (<i>n</i> = 6, 40%).</p><p>Treatment with ESAs was not associated with major side effects: our main concern, deep venous thrombosis (DVT), was reported in the clinical course of 19 patients (58%), but only in four cases occurred during ESA treatment (21%); all these patients had high CRP levels at the beginning of ESA therapy, suggesting a baseline inflammatory state.</p><p>At last follow-up, 24 patients were still alive (75%), with a median follow-up of 22.8 months (IQR: 14.3–34 months). We observed eight deaths during follow-up. Interestingly, 78 patients who died had either LOR or NR and were transfusion dependent at the time of ESAs discontinuation.</p><p>No standardized treatment exists for macrocytic anaemia in VEXAS patients, reported in over 90% of cases.<span><sup>9</sup></span> Management typically involves chronic red blood cell transfusions, associated with iron overload and impaired quality of life. The aetiology of anaemia in VEXAS remains unclear, but both systemic inflammation and the role of <i>UBA1</i> mutation in erythroblast differentiation are invoked: Recent data suggest that <i>UBA1</i> mutations lead to erythroblastopenia, with a skewed contribution from <i>UBA1</i> wild-type erythroid progenitors.<span><sup>10</sup></span></p><p>ESAs are standard therapies for low-risk anaemic MDS.<span><sup>11</sup></span> In this study, we analysed 32 VEXAS patients treated with ESAs, mostly with coexisting MDS. HI-E was achieved in 59% of patients, aligning with response rates in lower risk MDS.<span><sup>8</sup></span> Response correlated with LTB and low baseline EPO levels, consistent with the Hellström-Lindberg score.<span><sup>8</sup></span> However, the median duration of response in our patients was significantly shorter (13 months, IQR: 7.5–26.5). The cohort's heterogeneity and prior treatments, unlike typical non-inflamed MDS populations, limit conclusions on ESAs' impact on VEXAS disease activity.</p><p>DVT is a well-known feature of VEXAS (up to 35% of cases) due to chronic autoinflammation and increased cytokine release, leading to hypercoagulability and endothelial dysfunction.<span><sup>12</sup></span></p><p>In our cohort, 59% of patients had had a history of thrombosis, with only four events occurring during ESA treatment; these results are in line with preliminary observations from other groups on the reasonable safety profile of ESAs in VEXAS.<span><sup>13</sup></span> The generally low thrombotic rate may reflect concurrent anti-inflammatory treatment with glucocorticoids or biological disease-modifying antirheumatic drugs (bDMARDs). ESA response did not correlate with <i>UBA1</i> clonal burden, consistent with evidence that clone size is only affected by clone-targeting therapies (e.g. azacitidine<span><sup>14</sup></span> or allo-HCT<span><sup>15</sup></span>). Notably, mortality occurred mainly in patients who were transfusion dependent due to ESA failure or loss of response, highlighting anaemia as a potential prognostic factor.</p><p>Our results support the efficacy and safety of ESAs in VEXAS patients. Prospective studies with therapies like ESAs or Luspatercept, which have recently proven useful in improving anaemia in low-risk MDS,<span><sup>16</sup></span> are warranted to identify the best treatment options for anaemia in VEXAS.</p><p>ED, CC, GF, FM and CG supervised the project and wrote the manuscript. All the remaining authors contributed with patient's data and samples and participated in the discussion, read, edited and approved the final version of this manuscript, being accountable for all aspects of the work.</p><p>The authors declare no competing financial interests.</p><p>Chart review for clinical data was performed in accordance with the protocols set by the institutional review boards of each participating institution and the Declaration of Helsinki.</p>","PeriodicalId":135,"journal":{"name":"British Journal of Haematology","volume":"207 1","pages":"273-277"},"PeriodicalIF":3.8000,"publicationDate":"2025-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bjh.20157","citationCount":"0","resultStr":"{\"title\":\"Erythroid-stimulating agents in VEXAS syndrome: A retrospective study from an Italian multicentre cohort\",\"authors\":\"E. Diral, C. Campochiaro, G. Furnari, F. Moretti, J. Ferrari, C. Elena, G. Battipaglia, S. Barbato, A. Vitale, M. Frigeni, F. Crisafulli, M. Frassi, C. Papayannidis, A. D. Romagnoli, C. Cattaneo, A. D'Ambrosio, E. Morsia, P. Musto, G. Rivoli, M. E. Dragani, C. Toffalori, G. M. Bergonzi, G. Scorpio, A. Tomelleri, M. T. Voso, C. Gurnari, L. Vago, L. Dagna, F. Ciceri\",\"doi\":\"10.1111/bjh.20157\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>VEXAS (Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic) syndrome is an autoinflammatory disorder caused by somatic mutations in the <i>UBA1</i> gene, resulting in impaired protein ubiquitylation.<span><sup>1</sup></span> Patients present with inflammatory symptoms and a variety of haematological conditions, including plasma cell dyscrasias and cytopenias,<span><sup>2</sup></span> mostly macrocytic anaemia.<span><sup>1</sup></span> Accordingly, patients can be categorized as per the WHO 2022 criteria into ICUS (Idiopathic Cytopenia of Uncertain Significance), CCUS (Clonal Cytopenia of Uncertain Significance) or MDS (Myelodysplastic Neoplasia) in up to 50% of patients.<span><sup>3</sup></span></p><p>Currently, no standard therapy is available for VEXAS. For patients with moderate-to-severe anaemia, transfusions and erythropoiesis-stimulating agents (ESAs) are used, borrowing practices from lower risk MDS with the goal of improving quality of life. In this retrospective study, we analyse the use of ESAs in VEXAS patients, their efficacy and safety.</p><p>This study was conducted through a national survey sent to centres known to be involved in the clinical management of VEXAS patients. An electronic questionnaire was distributed from September to December 2024. We included patients with a confirmed diagnosis of VEXAS by <i>UBA1</i> testing receiving ESA treatment. Centres were queried for relevant data concerning clinical manifestations of VEXAS, haematological conditions as per the WHO 2022<span><sup>4</sup></span> and ESA treatment. Response to ESAs was evaluated after at least 16 weeks according to the IWG 2018 criteria.<span><sup>5</sup></span> MDS risk assessment was assigned as per Revised International Prognostic Scoring System (IPSS-R) and its molecular (IPSS-M) version. <i>UBA1</i> variant allele frequency (VAF) was determined by digital droplet PCR (ddPCR).<span><sup>6</sup></span> The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.</p><p>Patients' features were summarized using descriptive statistics. Quantitative variables were expressed as medians and interquartile range (IQR). Qualitative variables were expressed as numbers and percentages. Univariable and multivariable logistic regression analyses with odds ratio (OR) and 95% confidence interval (CI) were applied to analyse the ability of baseline features to predict response to ESA. Analysed variables included: underlying haematological condition; IPSS-R and IPSS-M risk categories; endogenous EPO levels; baseline haemoglobin levels; transfusion dependence; UBA1 clone size; and baseline CRP levels. Analyses, graphs and data visualizations were generated using Microsoft Excel and Graphpad Prism softwares.</p><p>Overall, 32 male patients received ESA treatment in 13 centres. Median age at VEXAS diagnosis was 66 years (IQR: 64–73). Clinical manifestations are reported in Table 1. Regarding <i>UBA1</i> variants, all patients harboured the canonical p.Met41Thr (<i>n</i> = 16, 50%), p.Met41Val (<i>n</i> = 12, 38%) and p.Met41Leu (<i>n</i> = 4, 12%) mutations. Next-generation sequencing (NGS) before ESA initiation was available for 25 patients (78%) and detected additional somatic myeloid mutations in 72% of cases (median of 1 mutation per patient, IQR: 0–1). The most frequently mutated genes belonged to the DTA triad<span><sup>7</sup></span>: DNMT3A, TET2, ASXL1 found in 72% (<i>n</i> = 13), 17% (<i>n</i> = 3) and 22% (<i>n</i> = 4) of cases respectively (Figure 1A). Twenty-six patients (82%) had concomitant MDS (Figure 1B), predominantly MDS-LB according to the WHO 2022 (73%, <i>n</i> = 19, Figure 1C). ICUS and CCUS were diagnosed in 3 (9%) patients each. The majority of MDS belonged to lower risk categories of IPSS-R/M scoring systems (IPSS-R <i>n</i> = 22, 85%; IPSS-M <i>n</i> = 17, 65%; Figure 1D).</p><p>Twenty-eight patients (88%) had symptomatic anaemia at baseline, with median haemoglobin levels of 91 g/L (range: 74–10). Of them, 20 (63%) were transfusion-dependent: 13 had a low transfusion burden (LTB, 41.9%) and 7 (35%) a high transfusion burden (HTB) (Figure 1E).</p><p>Before ESA initiation, <i>UBA1</i> VAF determination by ddPCR was available in 17 patients (53%, median 75%, IQR: 57.84–86.95), median C-reactive protein (CRP) levels were 15.8 mg/L (IQR: 7.6–53) and endogenous erythropoietin (EPO) levels were 124 mU/mL (IQR: 57.4–201). Concomitant treatments are shown in Figure 1F: 97% (<i>N</i> = 30) of patients were receiving VEXAS-directed anti-inflammatory therapy. Azacitidine was administered concomitantly with ESAs in two cases: Both achieved haematological improvement–erythroid (HI-E) and transfusion independence after azacitidine initiation. In these two cases, haemoglobin improvement likely reflects the effect of the hypomethylating agent, though UBA1 clone monitoring data are lacking.</p><p>Overall, 20 (63%) and 9 (28%) patients received epoetin alpha or epoetin zeta, at a median starting dose of 40 000 IU weekly (IQR: 30 000–40 000). Three patients received darbepoetin. In 11 patients (34%), the ESA dose was increased to the maximum of 80 000 IU weekly. Five of 11 patients (45%) receiving the maximum recommended dose eventually achieved a response. The median duration of exposure to ESAs was 41.3 weeks (range: 0.3–205.7) in our cohort.</p><p>HI-E was achieved in 19 patients (59%), with all responders obtaining transfusion independence (Figure S1). Responding patients were mostly LTB (58%, <i>n</i> = 11) or non-transfusion dependent (NTD, 37%, <i>n</i> = 7) and had lower median endogenous EPO levels (123 mU/mL, IQR: 51.2–177.7) compared to patients who had no response (NR; 124 mU/mL, IQR: 67–200), consistent with the Hellström-Lindberg score.<span><sup>8</sup></span> Of the seven HTB patients, only 1 (14%) obtained a major HI-E response, lasting 27 months. Six of 13 non-responding patients (46%) received ESAs full dose (80 000 IU/week) and the duration of exposure to ESA in these patients was >8 weeks. In non-responders, median CRP level changed from 14.5 mg/L prior to therapy (IQR: 9.9–55.9) to 21.3 mg/L at discontinuation (IQR: 10.78–54.4). In patients who lost response, median CRP level was 19.3 mg/L at baseline (IQR: 3–161.95) compared to 19.5 mg/L at ESA discontinuation.</p><p>At multivariate analysis, endogenous EPO was the only variable associated with the probability of response to ESAs (OR: 0.985; 95% CI = 0.972–0.999; <i>p</i> = 0.033); the type of <i>UBA1</i> mutation, as well as concurrent somatic myeloid mutations, did not correlate with response.</p><p>Median duration of response to ESAs was 13 months (IQR: 7.5–26.5). Six patients who initially achieved HI-E lost response (LOR, 31%) after a median of 14 months (IQR: 9.5–27.75). Median follow-up for responding patients was 15.6 months (IQR: 7.9–27.7 months).</p><p>Overall, 15/19 patients (79%) were transfusion dependent at ESA discontinuation due to NR or LOR. ddPCR for UBA1 was available at ESA discontinuation for four patients, and no significant changes were observed (Figure 1G).</p><p>For patients who had NR or LOR, subsequent treatments after ESA discontinuation included biological disease-modifying anti-rheumatic drugs (bDMARDs, <i>n</i> = 6, 31%), azacitidine alone or in combination with bDMARDSs (<i>n</i> = 4, 21%), allogeneic haematopoietic cell transplantation (allo-HCT, <i>n</i> = 6, 31%) or supportive care (<i>n</i> = 2, 10%), including red blood cell transfusions. Patients with long-lasting response to ESAs also underwent allo-HCT (<i>n</i> = 1) or azacitidine (<i>n</i> = 6, 40%).</p><p>Treatment with ESAs was not associated with major side effects: our main concern, deep venous thrombosis (DVT), was reported in the clinical course of 19 patients (58%), but only in four cases occurred during ESA treatment (21%); all these patients had high CRP levels at the beginning of ESA therapy, suggesting a baseline inflammatory state.</p><p>At last follow-up, 24 patients were still alive (75%), with a median follow-up of 22.8 months (IQR: 14.3–34 months). We observed eight deaths during follow-up. Interestingly, 78 patients who died had either LOR or NR and were transfusion dependent at the time of ESAs discontinuation.</p><p>No standardized treatment exists for macrocytic anaemia in VEXAS patients, reported in over 90% of cases.<span><sup>9</sup></span> Management typically involves chronic red blood cell transfusions, associated with iron overload and impaired quality of life. The aetiology of anaemia in VEXAS remains unclear, but both systemic inflammation and the role of <i>UBA1</i> mutation in erythroblast differentiation are invoked: Recent data suggest that <i>UBA1</i> mutations lead to erythroblastopenia, with a skewed contribution from <i>UBA1</i> wild-type erythroid progenitors.<span><sup>10</sup></span></p><p>ESAs are standard therapies for low-risk anaemic MDS.<span><sup>11</sup></span> In this study, we analysed 32 VEXAS patients treated with ESAs, mostly with coexisting MDS. HI-E was achieved in 59% of patients, aligning with response rates in lower risk MDS.<span><sup>8</sup></span> Response correlated with LTB and low baseline EPO levels, consistent with the Hellström-Lindberg score.<span><sup>8</sup></span> However, the median duration of response in our patients was significantly shorter (13 months, IQR: 7.5–26.5). The cohort's heterogeneity and prior treatments, unlike typical non-inflamed MDS populations, limit conclusions on ESAs' impact on VEXAS disease activity.</p><p>DVT is a well-known feature of VEXAS (up to 35% of cases) due to chronic autoinflammation and increased cytokine release, leading to hypercoagulability and endothelial dysfunction.<span><sup>12</sup></span></p><p>In our cohort, 59% of patients had had a history of thrombosis, with only four events occurring during ESA treatment; these results are in line with preliminary observations from other groups on the reasonable safety profile of ESAs in VEXAS.<span><sup>13</sup></span> The generally low thrombotic rate may reflect concurrent anti-inflammatory treatment with glucocorticoids or biological disease-modifying antirheumatic drugs (bDMARDs). ESA response did not correlate with <i>UBA1</i> clonal burden, consistent with evidence that clone size is only affected by clone-targeting therapies (e.g. azacitidine<span><sup>14</sup></span> or allo-HCT<span><sup>15</sup></span>). Notably, mortality occurred mainly in patients who were transfusion dependent due to ESA failure or loss of response, highlighting anaemia as a potential prognostic factor.</p><p>Our results support the efficacy and safety of ESAs in VEXAS patients. Prospective studies with therapies like ESAs or Luspatercept, which have recently proven useful in improving anaemia in low-risk MDS,<span><sup>16</sup></span> are warranted to identify the best treatment options for anaemia in VEXAS.</p><p>ED, CC, GF, FM and CG supervised the project and wrote the manuscript. All the remaining authors contributed with patient's data and samples and participated in the discussion, read, edited and approved the final version of this manuscript, being accountable for all aspects of the work.</p><p>The authors declare no competing financial interests.</p><p>Chart review for clinical data was performed in accordance with the protocols set by the institutional review boards of each participating institution and the Declaration of Helsinki.</p>\",\"PeriodicalId\":135,\"journal\":{\"name\":\"British Journal of Haematology\",\"volume\":\"207 1\",\"pages\":\"273-277\"},\"PeriodicalIF\":3.8000,\"publicationDate\":\"2025-05-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1111/bjh.20157\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"British Journal of Haematology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1111/bjh.20157\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"HEMATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"British Journal of Haematology","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1111/bjh.20157","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
Erythroid-stimulating agents in VEXAS syndrome: A retrospective study from an Italian multicentre cohort
VEXAS (Vacuoles, E1 Enzyme, X-linked, Autoinflammatory, Somatic) syndrome is an autoinflammatory disorder caused by somatic mutations in the UBA1 gene, resulting in impaired protein ubiquitylation.1 Patients present with inflammatory symptoms and a variety of haematological conditions, including plasma cell dyscrasias and cytopenias,2 mostly macrocytic anaemia.1 Accordingly, patients can be categorized as per the WHO 2022 criteria into ICUS (Idiopathic Cytopenia of Uncertain Significance), CCUS (Clonal Cytopenia of Uncertain Significance) or MDS (Myelodysplastic Neoplasia) in up to 50% of patients.3
Currently, no standard therapy is available for VEXAS. For patients with moderate-to-severe anaemia, transfusions and erythropoiesis-stimulating agents (ESAs) are used, borrowing practices from lower risk MDS with the goal of improving quality of life. In this retrospective study, we analyse the use of ESAs in VEXAS patients, their efficacy and safety.
This study was conducted through a national survey sent to centres known to be involved in the clinical management of VEXAS patients. An electronic questionnaire was distributed from September to December 2024. We included patients with a confirmed diagnosis of VEXAS by UBA1 testing receiving ESA treatment. Centres were queried for relevant data concerning clinical manifestations of VEXAS, haematological conditions as per the WHO 20224 and ESA treatment. Response to ESAs was evaluated after at least 16 weeks according to the IWG 2018 criteria.5 MDS risk assessment was assigned as per Revised International Prognostic Scoring System (IPSS-R) and its molecular (IPSS-M) version. UBA1 variant allele frequency (VAF) was determined by digital droplet PCR (ddPCR).6 The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Patients' features were summarized using descriptive statistics. Quantitative variables were expressed as medians and interquartile range (IQR). Qualitative variables were expressed as numbers and percentages. Univariable and multivariable logistic regression analyses with odds ratio (OR) and 95% confidence interval (CI) were applied to analyse the ability of baseline features to predict response to ESA. Analysed variables included: underlying haematological condition; IPSS-R and IPSS-M risk categories; endogenous EPO levels; baseline haemoglobin levels; transfusion dependence; UBA1 clone size; and baseline CRP levels. Analyses, graphs and data visualizations were generated using Microsoft Excel and Graphpad Prism softwares.
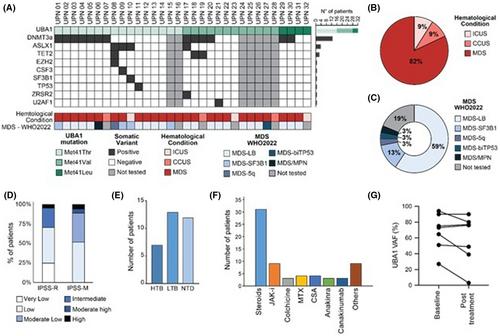
Overall, 32 male patients received ESA treatment in 13 centres. Median age at VEXAS diagnosis was 66 years (IQR: 64–73). Clinical manifestations are reported in Table 1. Regarding UBA1 variants, all patients harboured the canonical p.Met41Thr (n = 16, 50%), p.Met41Val (n = 12, 38%) and p.Met41Leu (n = 4, 12%) mutations. Next-generation sequencing (NGS) before ESA initiation was available for 25 patients (78%) and detected additional somatic myeloid mutations in 72% of cases (median of 1 mutation per patient, IQR: 0–1). The most frequently mutated genes belonged to the DTA triad7: DNMT3A, TET2, ASXL1 found in 72% (n = 13), 17% (n = 3) and 22% (n = 4) of cases respectively (Figure 1A). Twenty-six patients (82%) had concomitant MDS (Figure 1B), predominantly MDS-LB according to the WHO 2022 (73%, n = 19, Figure 1C). ICUS and CCUS were diagnosed in 3 (9%) patients each. The majority of MDS belonged to lower risk categories of IPSS-R/M scoring systems (IPSS-R n = 22, 85%; IPSS-M n = 17, 65%; Figure 1D).
Twenty-eight patients (88%) had symptomatic anaemia at baseline, with median haemoglobin levels of 91 g/L (range: 74–10). Of them, 20 (63%) were transfusion-dependent: 13 had a low transfusion burden (LTB, 41.9%) and 7 (35%) a high transfusion burden (HTB) (Figure 1E).
Before ESA initiation, UBA1 VAF determination by ddPCR was available in 17 patients (53%, median 75%, IQR: 57.84–86.95), median C-reactive protein (CRP) levels were 15.8 mg/L (IQR: 7.6–53) and endogenous erythropoietin (EPO) levels were 124 mU/mL (IQR: 57.4–201). Concomitant treatments are shown in Figure 1F: 97% (N = 30) of patients were receiving VEXAS-directed anti-inflammatory therapy. Azacitidine was administered concomitantly with ESAs in two cases: Both achieved haematological improvement–erythroid (HI-E) and transfusion independence after azacitidine initiation. In these two cases, haemoglobin improvement likely reflects the effect of the hypomethylating agent, though UBA1 clone monitoring data are lacking.
Overall, 20 (63%) and 9 (28%) patients received epoetin alpha or epoetin zeta, at a median starting dose of 40 000 IU weekly (IQR: 30 000–40 000). Three patients received darbepoetin. In 11 patients (34%), the ESA dose was increased to the maximum of 80 000 IU weekly. Five of 11 patients (45%) receiving the maximum recommended dose eventually achieved a response. The median duration of exposure to ESAs was 41.3 weeks (range: 0.3–205.7) in our cohort.
HI-E was achieved in 19 patients (59%), with all responders obtaining transfusion independence (Figure S1). Responding patients were mostly LTB (58%, n = 11) or non-transfusion dependent (NTD, 37%, n = 7) and had lower median endogenous EPO levels (123 mU/mL, IQR: 51.2–177.7) compared to patients who had no response (NR; 124 mU/mL, IQR: 67–200), consistent with the Hellström-Lindberg score.8 Of the seven HTB patients, only 1 (14%) obtained a major HI-E response, lasting 27 months. Six of 13 non-responding patients (46%) received ESAs full dose (80 000 IU/week) and the duration of exposure to ESA in these patients was >8 weeks. In non-responders, median CRP level changed from 14.5 mg/L prior to therapy (IQR: 9.9–55.9) to 21.3 mg/L at discontinuation (IQR: 10.78–54.4). In patients who lost response, median CRP level was 19.3 mg/L at baseline (IQR: 3–161.95) compared to 19.5 mg/L at ESA discontinuation.
At multivariate analysis, endogenous EPO was the only variable associated with the probability of response to ESAs (OR: 0.985; 95% CI = 0.972–0.999; p = 0.033); the type of UBA1 mutation, as well as concurrent somatic myeloid mutations, did not correlate with response.
Median duration of response to ESAs was 13 months (IQR: 7.5–26.5). Six patients who initially achieved HI-E lost response (LOR, 31%) after a median of 14 months (IQR: 9.5–27.75). Median follow-up for responding patients was 15.6 months (IQR: 7.9–27.7 months).
Overall, 15/19 patients (79%) were transfusion dependent at ESA discontinuation due to NR or LOR. ddPCR for UBA1 was available at ESA discontinuation for four patients, and no significant changes were observed (Figure 1G).
For patients who had NR or LOR, subsequent treatments after ESA discontinuation included biological disease-modifying anti-rheumatic drugs (bDMARDs, n = 6, 31%), azacitidine alone or in combination with bDMARDSs (n = 4, 21%), allogeneic haematopoietic cell transplantation (allo-HCT, n = 6, 31%) or supportive care (n = 2, 10%), including red blood cell transfusions. Patients with long-lasting response to ESAs also underwent allo-HCT (n = 1) or azacitidine (n = 6, 40%).
Treatment with ESAs was not associated with major side effects: our main concern, deep venous thrombosis (DVT), was reported in the clinical course of 19 patients (58%), but only in four cases occurred during ESA treatment (21%); all these patients had high CRP levels at the beginning of ESA therapy, suggesting a baseline inflammatory state.
At last follow-up, 24 patients were still alive (75%), with a median follow-up of 22.8 months (IQR: 14.3–34 months). We observed eight deaths during follow-up. Interestingly, 78 patients who died had either LOR or NR and were transfusion dependent at the time of ESAs discontinuation.
No standardized treatment exists for macrocytic anaemia in VEXAS patients, reported in over 90% of cases.9 Management typically involves chronic red blood cell transfusions, associated with iron overload and impaired quality of life. The aetiology of anaemia in VEXAS remains unclear, but both systemic inflammation and the role of UBA1 mutation in erythroblast differentiation are invoked: Recent data suggest that UBA1 mutations lead to erythroblastopenia, with a skewed contribution from UBA1 wild-type erythroid progenitors.10
ESAs are standard therapies for low-risk anaemic MDS.11 In this study, we analysed 32 VEXAS patients treated with ESAs, mostly with coexisting MDS. HI-E was achieved in 59% of patients, aligning with response rates in lower risk MDS.8 Response correlated with LTB and low baseline EPO levels, consistent with the Hellström-Lindberg score.8 However, the median duration of response in our patients was significantly shorter (13 months, IQR: 7.5–26.5). The cohort's heterogeneity and prior treatments, unlike typical non-inflamed MDS populations, limit conclusions on ESAs' impact on VEXAS disease activity.
DVT is a well-known feature of VEXAS (up to 35% of cases) due to chronic autoinflammation and increased cytokine release, leading to hypercoagulability and endothelial dysfunction.12
In our cohort, 59% of patients had had a history of thrombosis, with only four events occurring during ESA treatment; these results are in line with preliminary observations from other groups on the reasonable safety profile of ESAs in VEXAS.13 The generally low thrombotic rate may reflect concurrent anti-inflammatory treatment with glucocorticoids or biological disease-modifying antirheumatic drugs (bDMARDs). ESA response did not correlate with UBA1 clonal burden, consistent with evidence that clone size is only affected by clone-targeting therapies (e.g. azacitidine14 or allo-HCT15). Notably, mortality occurred mainly in patients who were transfusion dependent due to ESA failure or loss of response, highlighting anaemia as a potential prognostic factor.
Our results support the efficacy and safety of ESAs in VEXAS patients. Prospective studies with therapies like ESAs or Luspatercept, which have recently proven useful in improving anaemia in low-risk MDS,16 are warranted to identify the best treatment options for anaemia in VEXAS.
ED, CC, GF, FM and CG supervised the project and wrote the manuscript. All the remaining authors contributed with patient's data and samples and participated in the discussion, read, edited and approved the final version of this manuscript, being accountable for all aspects of the work.
The authors declare no competing financial interests.
Chart review for clinical data was performed in accordance with the protocols set by the institutional review boards of each participating institution and the Declaration of Helsinki.
期刊介绍:
The British Journal of Haematology publishes original research papers in clinical, laboratory and experimental haematology. The Journal also features annotations, reviews, short reports, images in haematology and Letters to the Editor.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


