Kai Cheng, Peng Wu, Wenbo Hu, Guotai Zhang, Shijie Guo, Sandong Guo and Yan Su
{"title":"二维层状BP/群- iv单硫族范德瓦尔斯异质结构:电子结构、能带取向和载流子动力学","authors":"Kai Cheng, Peng Wu, Wenbo Hu, Guotai Zhang, Shijie Guo, Sandong Guo and Yan Su","doi":"10.1039/D5CP00830A","DOIUrl":null,"url":null,"abstract":"<p >van der Waals heterostructures, composed of two-dimensional materials, have emerged as a promising platform for optoelectronic and photovoltaic applications. In this study, we constructed four vdW heterostructures, including BP/GeS, BP/GeSe, BP/SnS, and BP/SnSe, using BP and group-IV monochalcogenides as building blocks and investigated their potential for photovoltaic applications. First-principles calculations reveal that BP forms type II heterostructures with GeSe, SnS, and SnSe, while the BP/GeS system exhibits type I band alignment due to interfacial charge transfer. The structural stability of these heterostructures is confirmed by binding energy calculations, with the experimental synthesis of BP/SnSe providing additional validation. These heterostructures exhibit strong absorption in the infrared and visible regions, with predicted power conversion efficiencies ranging from 9.53% to 11.40%. Time-dependent density functional theory coupled with molecular dynamics simulations demonstrates ultrafast charge transfer in BP/GeSe, with electron transfer times (<em>τ</em> ≈ 147 fs) and hole transfer times (<em>τ</em> ≈ 839 fs), and the slower hole transfer rate is attributed to the limited mixing of higher energy levels and a coherent coupling mechanism. This work advances the fundamental understanding of BP/group-IV monochalcogenide van der Waals heterostructures and provides a robust theoretical foundation for their application in next-generation photovoltaic devices. Future research should focus on experimental validation, strain and stacking effects, and detailed device integration studies to fully harness their potential.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 22","pages":" 12060-12068"},"PeriodicalIF":2.9000,"publicationDate":"2025-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"2D layered BP/group-IV monochalcogenide van der Waals heterostructures for photovoltaics: electronic structure, band alignment, and carrier dynamics†\",\"authors\":\"Kai Cheng, Peng Wu, Wenbo Hu, Guotai Zhang, Shijie Guo, Sandong Guo and Yan Su\",\"doi\":\"10.1039/D5CP00830A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >van der Waals heterostructures, composed of two-dimensional materials, have emerged as a promising platform for optoelectronic and photovoltaic applications. In this study, we constructed four vdW heterostructures, including BP/GeS, BP/GeSe, BP/SnS, and BP/SnSe, using BP and group-IV monochalcogenides as building blocks and investigated their potential for photovoltaic applications. First-principles calculations reveal that BP forms type II heterostructures with GeSe, SnS, and SnSe, while the BP/GeS system exhibits type I band alignment due to interfacial charge transfer. The structural stability of these heterostructures is confirmed by binding energy calculations, with the experimental synthesis of BP/SnSe providing additional validation. These heterostructures exhibit strong absorption in the infrared and visible regions, with predicted power conversion efficiencies ranging from 9.53% to 11.40%. Time-dependent density functional theory coupled with molecular dynamics simulations demonstrates ultrafast charge transfer in BP/GeSe, with electron transfer times (<em>τ</em> ≈ 147 fs) and hole transfer times (<em>τ</em> ≈ 839 fs), and the slower hole transfer rate is attributed to the limited mixing of higher energy levels and a coherent coupling mechanism. This work advances the fundamental understanding of BP/group-IV monochalcogenide van der Waals heterostructures and provides a robust theoretical foundation for their application in next-generation photovoltaic devices. Future research should focus on experimental validation, strain and stacking effects, and detailed device integration studies to fully harness their potential.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 22\",\"pages\":\" 12060-12068\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-05-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00830a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00830a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
2D layered BP/group-IV monochalcogenide van der Waals heterostructures for photovoltaics: electronic structure, band alignment, and carrier dynamics†
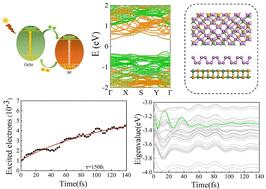
van der Waals heterostructures, composed of two-dimensional materials, have emerged as a promising platform for optoelectronic and photovoltaic applications. In this study, we constructed four vdW heterostructures, including BP/GeS, BP/GeSe, BP/SnS, and BP/SnSe, using BP and group-IV monochalcogenides as building blocks and investigated their potential for photovoltaic applications. First-principles calculations reveal that BP forms type II heterostructures with GeSe, SnS, and SnSe, while the BP/GeS system exhibits type I band alignment due to interfacial charge transfer. The structural stability of these heterostructures is confirmed by binding energy calculations, with the experimental synthesis of BP/SnSe providing additional validation. These heterostructures exhibit strong absorption in the infrared and visible regions, with predicted power conversion efficiencies ranging from 9.53% to 11.40%. Time-dependent density functional theory coupled with molecular dynamics simulations demonstrates ultrafast charge transfer in BP/GeSe, with electron transfer times (τ ≈ 147 fs) and hole transfer times (τ ≈ 839 fs), and the slower hole transfer rate is attributed to the limited mixing of higher energy levels and a coherent coupling mechanism. This work advances the fundamental understanding of BP/group-IV monochalcogenide van der Waals heterostructures and provides a robust theoretical foundation for their application in next-generation photovoltaic devices. Future research should focus on experimental validation, strain and stacking effects, and detailed device integration studies to fully harness their potential.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


