{"title":"从第一性原理计算Al对V储氢性能的影响","authors":"Jutao Hu, Xiaoqing Li and Stephan Schönecker","doi":"10.1039/D5CP00266D","DOIUrl":null,"url":null,"abstract":"<p >Vanadium-based materials have great potential for advancing novel hydrogen storage technology. To address the limited gravimetric hydrogen storage capacity of V, incorporating light alloying elements has been proposed. In this study, the hydrogen storage capacities of V<small><sub>1−<em>x</em></sub></small>Al<small><sub><em>x</em></sub></small> (<em>x</em> = 0, 0.1, 0.2, 0.3, and 0.4) solid solutions are investigated by employing first-principles calculations. Our results indicate that both the stability and hydrogen storage capacity of V<small><sub>1−<em>x</em></sub></small>Al<small><sub><em>x</em></sub></small> hydrides decrease with an increase in Al content due to a reduction of chemical contribution, consistent with experimental results. The chemical bond analysis, Bader charge, and projected density of states investigation reveal that the Al–H antibonding states appear at the Fermi level and net H–H antibonding states surrounding Al form due to the transfer of excessive electrons from Al to H. To further explore the relationship between chemical bonding and desorption enthalpy, over 20 face-centered cubic (FCC) metal dihydrides are selected. It is found that the desorption enthalpies correlate weakly with the metal–hydrogen (M–H) bond strength and positively with M–H antibonding states below the Fermi level. Our study reveals the mechanism of interactions between chemical bonds and hydrogen storage properties in metal hydrides, providing valuable insights for the future design of hydrogen storage materials.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 20","pages":" 10815-10825"},"PeriodicalIF":2.9000,"publicationDate":"2025-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The effects of Al on the hydrogen storage properties of V from first-principles calculations†\",\"authors\":\"Jutao Hu, Xiaoqing Li and Stephan Schönecker\",\"doi\":\"10.1039/D5CP00266D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Vanadium-based materials have great potential for advancing novel hydrogen storage technology. To address the limited gravimetric hydrogen storage capacity of V, incorporating light alloying elements has been proposed. In this study, the hydrogen storage capacities of V<small><sub>1−<em>x</em></sub></small>Al<small><sub><em>x</em></sub></small> (<em>x</em> = 0, 0.1, 0.2, 0.3, and 0.4) solid solutions are investigated by employing first-principles calculations. Our results indicate that both the stability and hydrogen storage capacity of V<small><sub>1−<em>x</em></sub></small>Al<small><sub><em>x</em></sub></small> hydrides decrease with an increase in Al content due to a reduction of chemical contribution, consistent with experimental results. The chemical bond analysis, Bader charge, and projected density of states investigation reveal that the Al–H antibonding states appear at the Fermi level and net H–H antibonding states surrounding Al form due to the transfer of excessive electrons from Al to H. To further explore the relationship between chemical bonding and desorption enthalpy, over 20 face-centered cubic (FCC) metal dihydrides are selected. It is found that the desorption enthalpies correlate weakly with the metal–hydrogen (M–H) bond strength and positively with M–H antibonding states below the Fermi level. Our study reveals the mechanism of interactions between chemical bonds and hydrogen storage properties in metal hydrides, providing valuable insights for the future design of hydrogen storage materials.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 20\",\"pages\":\" 10815-10825\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-05-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00266d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00266d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
The effects of Al on the hydrogen storage properties of V from first-principles calculations†
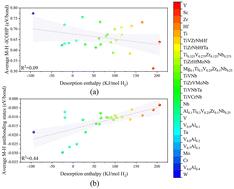
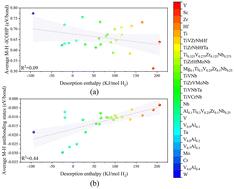
Vanadium-based materials have great potential for advancing novel hydrogen storage technology. To address the limited gravimetric hydrogen storage capacity of V, incorporating light alloying elements has been proposed. In this study, the hydrogen storage capacities of V1−xAlx (x = 0, 0.1, 0.2, 0.3, and 0.4) solid solutions are investigated by employing first-principles calculations. Our results indicate that both the stability and hydrogen storage capacity of V1−xAlx hydrides decrease with an increase in Al content due to a reduction of chemical contribution, consistent with experimental results. The chemical bond analysis, Bader charge, and projected density of states investigation reveal that the Al–H antibonding states appear at the Fermi level and net H–H antibonding states surrounding Al form due to the transfer of excessive electrons from Al to H. To further explore the relationship between chemical bonding and desorption enthalpy, over 20 face-centered cubic (FCC) metal dihydrides are selected. It is found that the desorption enthalpies correlate weakly with the metal–hydrogen (M–H) bond strength and positively with M–H antibonding states below the Fermi level. Our study reveals the mechanism of interactions between chemical bonds and hydrogen storage properties in metal hydrides, providing valuable insights for the future design of hydrogen storage materials.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


