{"title":"一些三价液态稀土金属熔点的静态、动态和电子性质:从头算和神经网络模拟。","authors":"Beatriz G. del Rio and Luis E. González","doi":"10.1039/D5CP00765H","DOIUrl":null,"url":null,"abstract":"<p >We report a study on several static and dynamic properties of the early trivalent liquid rare-earth metals at thermodynamic conditions near their respective melting points. It has been performed by resorting to machine learning (ML) techniques, in which the associated neural network-based interatomic potentials were derived from <em>ab initio</em> molecular dynamics simulations within Hubbard-corrected density functional theory. We report the results obtained for the static structural properties, including an analysis of the local short-range order. Single-particle and collective dynamic properties have also been obtained, from which transport coefficients and wavevector-dependent dispersion relations are evaluated. The results show a quite homogeneous behavior of the structural, dynamic, and transport properties throughout the series. The electronic properties have been obtained from the <em>ab initio</em> simulations, and show important discrepancies with respect to the low temperature solids, portraying a more band-like picture of the 4f states in the liquid.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 21","pages":" 11289-11299"},"PeriodicalIF":2.9000,"publicationDate":"2025-05-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00765h?page=search","citationCount":"0","resultStr":"{\"title\":\"Static, dynamic and electronic properties of some trivalent liquid rare earth metals near melting: ab initio and neural network simulations\",\"authors\":\"Beatriz G. del Rio and Luis E. González\",\"doi\":\"10.1039/D5CP00765H\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We report a study on several static and dynamic properties of the early trivalent liquid rare-earth metals at thermodynamic conditions near their respective melting points. It has been performed by resorting to machine learning (ML) techniques, in which the associated neural network-based interatomic potentials were derived from <em>ab initio</em> molecular dynamics simulations within Hubbard-corrected density functional theory. We report the results obtained for the static structural properties, including an analysis of the local short-range order. Single-particle and collective dynamic properties have also been obtained, from which transport coefficients and wavevector-dependent dispersion relations are evaluated. The results show a quite homogeneous behavior of the structural, dynamic, and transport properties throughout the series. The electronic properties have been obtained from the <em>ab initio</em> simulations, and show important discrepancies with respect to the low temperature solids, portraying a more band-like picture of the 4f states in the liquid.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 21\",\"pages\":\" 11289-11299\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-05-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00765h?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00765h\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00765h","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Static, dynamic and electronic properties of some trivalent liquid rare earth metals near melting: ab initio and neural network simulations
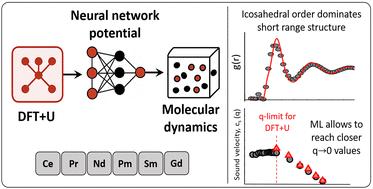
We report a study on several static and dynamic properties of the early trivalent liquid rare-earth metals at thermodynamic conditions near their respective melting points. It has been performed by resorting to machine learning (ML) techniques, in which the associated neural network-based interatomic potentials were derived from ab initio molecular dynamics simulations within Hubbard-corrected density functional theory. We report the results obtained for the static structural properties, including an analysis of the local short-range order. Single-particle and collective dynamic properties have also been obtained, from which transport coefficients and wavevector-dependent dispersion relations are evaluated. The results show a quite homogeneous behavior of the structural, dynamic, and transport properties throughout the series. The electronic properties have been obtained from the ab initio simulations, and show important discrepancies with respect to the low temperature solids, portraying a more band-like picture of the 4f states in the liquid.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


