Krishna Gopal Chattaraj, Joana Ferreira, Allan S. Myerson and Bernhardt L. Trout
{"title":"通过机器学习研究抗原结合片段结晶的结构生物物理特征","authors":"Krishna Gopal Chattaraj, Joana Ferreira, Allan S. Myerson and Bernhardt L. Trout","doi":"10.1039/D4ME00187G","DOIUrl":null,"url":null,"abstract":"<p >Antibody-based therapeutics continue to be an important pharmaceutical development modality. Crystallization of antibodies is important for structural characterization, but in addition has the potential for use as a separation method and for use as a dosage form. Nevertheless, bringing about controlled crystallization of an antibody remains a challenging task due to its large size, high degree of segmental flexibility, and the intricacy of all the occurring interactions (<em>e.g.</em>, protein–protein interactions, protein–solvent interactions, <em>etc.</em>). Methods to predict important contact sites could help to develop such crystallization methods. However, limited data and understanding have hitherto not allowed the development of such robust methods. This study employs machine learning combined with <em>in silico</em> modelling of crystal structures using available experimental structures to identify the crucial physicochemical features necessary for successful antibody crystallization in an attempt to remedy that gap. The developed method can with good accuracy distinguish crystal-site residues from non-crystal-site residues. A set of 510 descriptors is utilized to characterize each residue, which is treated as a distinct data point. Moreover, new algorithms have been developed to design novel descriptors that improve the model's predictive capabilities. Fragment antigen-binding (Fab) regions are investigated due to the scarcity of full-length monoclonal antibodies (mAbs) crystal structures. The current findings show that the extreme gradient boosting (XGBoost) algorithm effectively identifies crystal site residues, as evidenced by an AUPRC value that is more than 3-fold higher than that of the baseline model. The top-ranked descriptors indicate that crystal-site residues are primarily characterized by solvent-exposed residues with high spatial aggregation propensity (SAP), signifying hydrophobic patches, and their immediate surface-exposed neighbors. Moreover, these high SAP residues are often surrounded by other solvent-exposed residues that are either polar, charged, or both. In contrast, residues not involved in crystal interfaces generally lack these essential features, though some might be excluded due to specific crystal lattice arrangements. Additionally, reducing the feature set from 510 to the top 15% in the XGBoost model yields similar performance while significantly simplifying the model.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 5","pages":" 377-393"},"PeriodicalIF":3.2000,"publicationDate":"2025-03-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/me/d4me00187g?page=search","citationCount":"0","resultStr":"{\"title\":\"Investigating structural biophysical features for antigen-binding fragment crystallization via machine learning†\",\"authors\":\"Krishna Gopal Chattaraj, Joana Ferreira, Allan S. Myerson and Bernhardt L. Trout\",\"doi\":\"10.1039/D4ME00187G\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Antibody-based therapeutics continue to be an important pharmaceutical development modality. Crystallization of antibodies is important for structural characterization, but in addition has the potential for use as a separation method and for use as a dosage form. Nevertheless, bringing about controlled crystallization of an antibody remains a challenging task due to its large size, high degree of segmental flexibility, and the intricacy of all the occurring interactions (<em>e.g.</em>, protein–protein interactions, protein–solvent interactions, <em>etc.</em>). Methods to predict important contact sites could help to develop such crystallization methods. However, limited data and understanding have hitherto not allowed the development of such robust methods. This study employs machine learning combined with <em>in silico</em> modelling of crystal structures using available experimental structures to identify the crucial physicochemical features necessary for successful antibody crystallization in an attempt to remedy that gap. The developed method can with good accuracy distinguish crystal-site residues from non-crystal-site residues. A set of 510 descriptors is utilized to characterize each residue, which is treated as a distinct data point. Moreover, new algorithms have been developed to design novel descriptors that improve the model's predictive capabilities. Fragment antigen-binding (Fab) regions are investigated due to the scarcity of full-length monoclonal antibodies (mAbs) crystal structures. The current findings show that the extreme gradient boosting (XGBoost) algorithm effectively identifies crystal site residues, as evidenced by an AUPRC value that is more than 3-fold higher than that of the baseline model. The top-ranked descriptors indicate that crystal-site residues are primarily characterized by solvent-exposed residues with high spatial aggregation propensity (SAP), signifying hydrophobic patches, and their immediate surface-exposed neighbors. Moreover, these high SAP residues are often surrounded by other solvent-exposed residues that are either polar, charged, or both. In contrast, residues not involved in crystal interfaces generally lack these essential features, though some might be excluded due to specific crystal lattice arrangements. Additionally, reducing the feature set from 510 to the top 15% in the XGBoost model yields similar performance while significantly simplifying the model.</p>\",\"PeriodicalId\":91,\"journal\":{\"name\":\"Molecular Systems Design & Engineering\",\"volume\":\" 5\",\"pages\":\" 377-393\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-03-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/me/d4me00187g?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Systems Design & Engineering\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/me/d4me00187g\",\"RegionNum\":3,\"RegionCategory\":\"工程技术\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/me/d4me00187g","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Investigating structural biophysical features for antigen-binding fragment crystallization via machine learning†
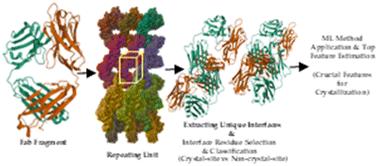
Antibody-based therapeutics continue to be an important pharmaceutical development modality. Crystallization of antibodies is important for structural characterization, but in addition has the potential for use as a separation method and for use as a dosage form. Nevertheless, bringing about controlled crystallization of an antibody remains a challenging task due to its large size, high degree of segmental flexibility, and the intricacy of all the occurring interactions (e.g., protein–protein interactions, protein–solvent interactions, etc.). Methods to predict important contact sites could help to develop such crystallization methods. However, limited data and understanding have hitherto not allowed the development of such robust methods. This study employs machine learning combined with in silico modelling of crystal structures using available experimental structures to identify the crucial physicochemical features necessary for successful antibody crystallization in an attempt to remedy that gap. The developed method can with good accuracy distinguish crystal-site residues from non-crystal-site residues. A set of 510 descriptors is utilized to characterize each residue, which is treated as a distinct data point. Moreover, new algorithms have been developed to design novel descriptors that improve the model's predictive capabilities. Fragment antigen-binding (Fab) regions are investigated due to the scarcity of full-length monoclonal antibodies (mAbs) crystal structures. The current findings show that the extreme gradient boosting (XGBoost) algorithm effectively identifies crystal site residues, as evidenced by an AUPRC value that is more than 3-fold higher than that of the baseline model. The top-ranked descriptors indicate that crystal-site residues are primarily characterized by solvent-exposed residues with high spatial aggregation propensity (SAP), signifying hydrophobic patches, and their immediate surface-exposed neighbors. Moreover, these high SAP residues are often surrounded by other solvent-exposed residues that are either polar, charged, or both. In contrast, residues not involved in crystal interfaces generally lack these essential features, though some might be excluded due to specific crystal lattice arrangements. Additionally, reducing the feature set from 510 to the top 15% in the XGBoost model yields similar performance while significantly simplifying the model.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


