深度学习增强的精确分子动力学钛势模型
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
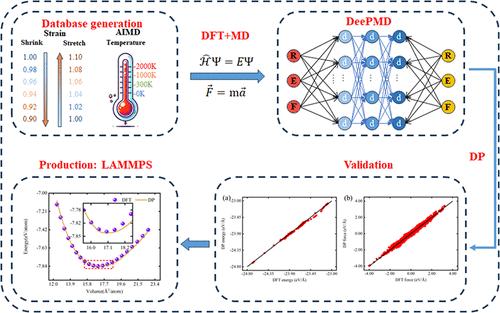
Deep potential (DP)是一种先进的基于深度学习的方法,它可以从密度泛函理论(DFT)计算中提取复杂的材料相互作用,以构建高精度的势能函数。在众多的材料体系中,过渡金属钛(Ti)由于其独特的电子结构和催化活性,在储能、转化和催化等领域受到了广泛的关注。然而,钛复杂的原子间相互作用给传统的势模型带来了挑战,使得准确的描述变得困难。为了解决这一挑战,我们开发了一种DP方法,能够有效地建立Ti分子动力学(MD)模拟所需的原子间势函数,从而提高模拟精度。本研究系统地比较了DP模型的预测结果与DFT的结果,表明DP模型在预测关键属性方面达到了DFT级别的精度。这些性能包括晶体结构特性、缺陷特性、表面和界面特性以及机械性能。α-相六方密排结构在1189 K时自动转变为β相体心立方结构,熔化温度为1864 K。这些数值与实验相变温度1209 K和熔点1941 K基本一致,证实了DP模型在高温条件下的准确性。温度升高后,Ti原子的自扩散系数达到2.51 × 10-10 m2/s,与其他面心立方金属的自扩散系数相当。该方法的成功应用为复杂材料的多尺度模拟和性能预测奠定了基础,具有推进新材料高通量筛选和精确建模的潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Deep Learning-Enhanced Titanium Potential Models for Accurate Molecular Dynamics
Deep potential (DP) is an advanced deep learning-based method that can extract complex material interactions from density functional theory (DFT) calculations to construct highly accurate potential energy functions. Among various material systems, the transition metal titanium (Ti) has garnered significant attention in fields such as energy storage and conversion and catalysis, due to its unique electronic structure and catalytic activity. However, the complex interatomic interactions of Ti pose challenges for traditional potential models, making accurate descriptions difficult. To address this challenge, we developed a DP method capable of effectively establishing the interatomic potential functions required for molecular dynamics (MD) simulations of Ti, thereby enhancing simulation accuracy. This study systematically compares the predictions of the DP model with results from DFT, demonstrating that the DP model achieves DFT-level accuracy in predicting key properties. These properties include crystal structural characteristics, defect properties, surface and interface properties, as well as mechanical properties. The α-phase Hexagonal Close-Packed structure automatically transforms into the β-phase Body-Centered Cubic structure at 1189 K, exhibiting a melting temperature of 1864 K. These values closely align with the experimental phase transition temperature of 1209 K and the melting point of 1941 K, confirming the DP model’s accuracy under high-temperature conditions. Increasing temperature enhances Ti atom diffusivity, with a self-diffusion coefficient observed at the melting temperature of 2.51 × 10–10 m2/s, matching those of other Face-Centered Cubic metals. The successful application of this method lays a foundation for multiscale simulations and performance predictions of complex materials, with potential to advance high-throughput screening and precise modeling of new materials.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


