{"title":"三层微溶剂化模型对Fe(III)/Fe(II)配合物氧化还原电位计算的系统改进","authors":"Hassan Harb and Rajeev Surendran Assary","doi":"10.1039/D5CP00454C","DOIUrl":null,"url":null,"abstract":"<p >Electrochemical transformations of metal ions in aqueous media are challenging to model accurately due to the dynamic solvation structure surrounding ions at different charge states. Predictive modeling at the atomistic scale is essential for understanding these solvation architectures but is often computationally prohibitive. In this contribution, we present a simple, fast, and accurate three-layer micro-solvation model to evaluate the redox potential of metal ions in aqueous solutions. Our model, developed and validated for Fe<small><sup>3+</sup></small>/Fe<small><sup>2+</sup></small> redox potentials, combines the DFT-based geometry optimizations of the octahedral Fe complex with two layers of explicit water molecules to capture solute–solvent interactions and an implicit solvation model to account for bulk solvent effects. This approach yields accurate predictions for Fe<small><sup>3+</sup></small>/Fe<small><sup>2+</sup></small> redox potentials in water, achieving errors of 0.02 V with ωB97X-V, 0.01 V with ωB97X-D3, 0.04 V with ωB97M-V, and 0.02 V with B3LYP-D3 functionals. We further demonstrate the generality of our model by applying it to additional metal complexes, including the challenging Fe(CN)<small><sub>6</sub></small><small><sup>3−/4−</sup></small> system, where our model successfully achieves close agreement with experimental values, with an error of 0.07 V and an average error of 0.21 V for all five systems. In summary, the presented simple solvation model has broad applicability and potential for enhancing computational efficiency in redox potential predictions across various chemical and industrial processes of metal ions.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 20","pages":" 10717-10729"},"PeriodicalIF":2.9000,"publicationDate":"2025-04-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00454c?page=search","citationCount":"0","resultStr":"{\"title\":\"Systematic improvement of redox potential calculation of Fe(iii)/Fe(ii) complexes using a three-layer micro-solvation model†\",\"authors\":\"Hassan Harb and Rajeev Surendran Assary\",\"doi\":\"10.1039/D5CP00454C\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Electrochemical transformations of metal ions in aqueous media are challenging to model accurately due to the dynamic solvation structure surrounding ions at different charge states. Predictive modeling at the atomistic scale is essential for understanding these solvation architectures but is often computationally prohibitive. In this contribution, we present a simple, fast, and accurate three-layer micro-solvation model to evaluate the redox potential of metal ions in aqueous solutions. Our model, developed and validated for Fe<small><sup>3+</sup></small>/Fe<small><sup>2+</sup></small> redox potentials, combines the DFT-based geometry optimizations of the octahedral Fe complex with two layers of explicit water molecules to capture solute–solvent interactions and an implicit solvation model to account for bulk solvent effects. This approach yields accurate predictions for Fe<small><sup>3+</sup></small>/Fe<small><sup>2+</sup></small> redox potentials in water, achieving errors of 0.02 V with ωB97X-V, 0.01 V with ωB97X-D3, 0.04 V with ωB97M-V, and 0.02 V with B3LYP-D3 functionals. We further demonstrate the generality of our model by applying it to additional metal complexes, including the challenging Fe(CN)<small><sub>6</sub></small><small><sup>3−/4−</sup></small> system, where our model successfully achieves close agreement with experimental values, with an error of 0.07 V and an average error of 0.21 V for all five systems. In summary, the presented simple solvation model has broad applicability and potential for enhancing computational efficiency in redox potential predictions across various chemical and industrial processes of metal ions.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 20\",\"pages\":\" 10717-10729\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-04-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00454c?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00454c\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00454c","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Systematic improvement of redox potential calculation of Fe(iii)/Fe(ii) complexes using a three-layer micro-solvation model†
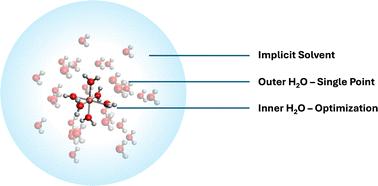
Electrochemical transformations of metal ions in aqueous media are challenging to model accurately due to the dynamic solvation structure surrounding ions at different charge states. Predictive modeling at the atomistic scale is essential for understanding these solvation architectures but is often computationally prohibitive. In this contribution, we present a simple, fast, and accurate three-layer micro-solvation model to evaluate the redox potential of metal ions in aqueous solutions. Our model, developed and validated for Fe3+/Fe2+ redox potentials, combines the DFT-based geometry optimizations of the octahedral Fe complex with two layers of explicit water molecules to capture solute–solvent interactions and an implicit solvation model to account for bulk solvent effects. This approach yields accurate predictions for Fe3+/Fe2+ redox potentials in water, achieving errors of 0.02 V with ωB97X-V, 0.01 V with ωB97X-D3, 0.04 V with ωB97M-V, and 0.02 V with B3LYP-D3 functionals. We further demonstrate the generality of our model by applying it to additional metal complexes, including the challenging Fe(CN)63−/4− system, where our model successfully achieves close agreement with experimental values, with an error of 0.07 V and an average error of 0.21 V for all five systems. In summary, the presented simple solvation model has broad applicability and potential for enhancing computational efficiency in redox potential predictions across various chemical and industrial processes of metal ions.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


