石墨表面水碰撞的化学动力学模拟
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
本研究采用分子动力学模拟方法,对D2O分子在高取向热解石墨表面的散射动力学进行了全面分析。MD模拟协议通过实验验证,其中只包含面内散射[Gibson, K. D., J. Chem.]。物理学报,2016,32 (1),393 - 393 [j]。然而,由于面外散射的显著贡献,面内结果无法捕捉整体动力学,这是难以测量的。在这项研究中,我们的模拟考虑了面内和面外散射,并表明中间入射角(~ 40°)最大限度地减少了水- hopg相互作用。通过分析内部拐点数目之间的关系,我们将入射角与散射分子的热化速率联系起来。我们的研究结果突出了MD模拟模拟复杂气体表面相互作用的能力,这些相互作用难以通过实验捕获,为未来更广泛的能量、温度、表面和多分子相互作用的研究提供了有效和准确的方法。本文章由计算机程序翻译,如有差异,请以英文原文为准。
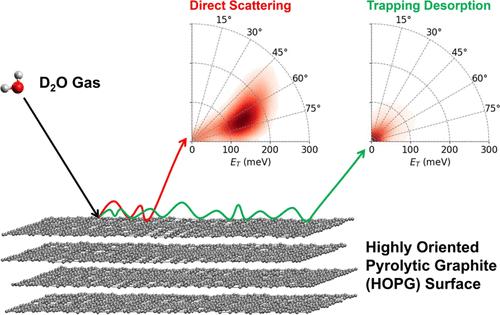
Chemical Dynamics Simulations of Water Collisions with a Graphite Surface
This study presents a comprehensive analysis of the scattering dynamics of D2O molecules on highly oriented pyrolytic graphite surfaces using molecular dynamics simulations. The MD simulation protocols are validated against experiments, which contain only in-plane scattering [Gibson, K. D., J. Chem. Phys. 2024, 160(1), 194705.10.1063/5.0205984]. However, the in-plane results fail to capture the overall dynamics due to the significant contribution of out-of-plane scattering, which is difficult to measure. In this study, our simulations consider both in- and out-of-plane scatterings and show that the intermediate incident angle (∼40°) minimizes water–HOPG interaction. By analyzing the relation between the number of internal turning points, we relate the incident angle to the rate of thermalization of the scattered-off molecules. Our findings highlight the capability of MD simulations to model complex gas–surface interactions that are difficult to capture experimentally, offering an effective and accurate method for future studies across a wider range of energies, temperatures, surfaces, and multimolecule interactions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


