基于python的机器学习过渡态加速计算自动化课程
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
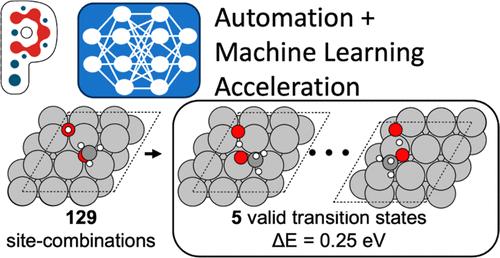
微动力学模型(mkm)广泛应用于计算多相催化领域,用于研究复杂的反应机理,使实验趋势合理化,并加速新型催化剂的合理设计。然而,构建这些模型需要计算昂贵且手动繁琐的密度泛函理论(DFT)计算,以确定MKM内每个基本反应的过渡态。为了解决这些挑战,我们展示了一种新的协议,该协议使用开源动力学工作流工具Pynta来自动化反应性机器学习潜力(rMLP)的迭代训练。具体来说,以银催化的甲醇部分氧化为原型,我们首先通过训练rMLP来加速所有53个反应的dft质量过渡态的并行计算来演示我们的工作流程,与仅dft策略相比,实现了7倍的加速。对我们培训课程的详细分析揭示了使用单一rMLP模型的自适应抽样方案同时描述MKM内所有反应的缺点。我们表明,这些限制可以通过使用多个rMLP模型的平衡“反应类”方法来克服,每个rMLP模型描述一个类似过渡状态的单一类别。最后,我们证明了基于python的工作流也与大型预训练的基础模型兼容。例如,通过微调在OC20数据集上训练的表现最好的图神经网络电位,我们观察到在识别过渡状态方面有令人印象深刻的20倍加速和89%的成功率。这项工作强调了将自动化工具与机器学习集成在一起以推进催化研究的协同潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Automated Pynta-Based Curriculum for ML-Accelerated Calculation of Transition States
Microkinetic models (MKMs) are widely used within the computational heterogeneous catalysis community to investigate complex reaction mechanisms, to rationalize experimental trends, and to accelerate the rational design of novel catalysts. However, constructing these models requires computationally expensive and manually tedious density functional theory (DFT) calculations for identifying transition states for each elementary reaction within the MKM. To address these challenges, we demonstrate a novel protocol that uses the open-source kinetics workflow tool Pynta to automate the iterative training of a reactive machine learning potential (rMLP). Specifically, using the silver-catalyzed partial oxidation of methanol as a prototypical example, we first demonstrate our workflow by training an rMLP to accelerate the parallel calculation of DFT-quality transition states for all 53 reactions, achieving a 7× speedup compared to a DFT-only strategy. Detailed analysis of our training curriculum reveals the shortcomings of using an adaptive sampling scheme with a single rMLP model to describe all reactions within the MKM simultaneously. We show that these limitations can be overcome using a balanced “reaction class” approach that uses multiple rMLP models, each describing a single class of similar transition states. Finally, we demonstrate that our Pynta-based workflow is also compatible with large pretrained foundational models. For example, by fine-tuning a top-performing graph neural network potential trained on the OC20 dataset, we observe an impressive 20× speedup with an 89% success rate in identifying transition states. This work highlights the synergistic potential of integrating automated tools with machine learning to advance catalysis research.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


