{"title":"酶替代治疗对不同严重程度的兄弟姐妹低磷酸酶血症的影响。","authors":"Junko Kanno, Tomohiro Nakagawa, Akinobu Miura, Hirohito Shima, Chisumi Sogi, Miki Kamimura, Ikuma Fujiwara, Kanako Tachikawa, Ryoko Hino, Toshimi Michigami, Atsuo Kikuchi","doi":"10.1297/cpe.2024-0084","DOIUrl":null,"url":null,"abstract":"<p><p>Hypophosphatasia (HPP) is a hereditary disorder characterized by impaired bone mineralization caused by decreased tissue-nonspecific alkaline phosphatase (TNSALP) activity. Specifically, HPP is caused by a loss-of-function variant in the <i>ALPL</i> gene encoding TNSALP. Although genotype-phenotype correlations have been described, phenotypic differences have been reported in patients with the same variants, even within families. The proband, a girl, was suspected to have in utero fractures of the long bones, suggestive of osteogenesis imperfecta. No respiratory impairment was observed after birth; however, the patient's serum alkaline phosphatase level was low. In addition, the patient's perinatal findings were consistent with those of perinatal benign HPP, although the bone symptoms subsequently worsened. The patient's brother, initially suspected to have odonto-HPP due to the premature loss of primary teeth, later developed compression fractures and extraosseous symptoms. Both patients had the same <i>ALPL</i> variants, c. 572A>G(;)1559del, p. Glu191Gly(;)Leu520ArgfsTer86; however, the severity of their conditions differed. Patients with HPP with identical genotypes in the same family may have varying severity levels of HPP. In this case report, both patients received enzyme replacement therapy (ERT), which improved the clinical symptoms. Therefore, for perinatal benign HPP, ERT should be considered if bone symptoms worsen. In addition, odonto-HPP should be closely monitored, and ERT should be considered if bone and extraosseous symptoms arise.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"34 2","pages":"137-143"},"PeriodicalIF":1.2000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11972865/pdf/","citationCount":"0","resultStr":"{\"title\":\"Effects of enzyme replacement therapy in sibling cases of hypophosphatasia of varying severities.\",\"authors\":\"Junko Kanno, Tomohiro Nakagawa, Akinobu Miura, Hirohito Shima, Chisumi Sogi, Miki Kamimura, Ikuma Fujiwara, Kanako Tachikawa, Ryoko Hino, Toshimi Michigami, Atsuo Kikuchi\",\"doi\":\"10.1297/cpe.2024-0084\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hypophosphatasia (HPP) is a hereditary disorder characterized by impaired bone mineralization caused by decreased tissue-nonspecific alkaline phosphatase (TNSALP) activity. Specifically, HPP is caused by a loss-of-function variant in the <i>ALPL</i> gene encoding TNSALP. Although genotype-phenotype correlations have been described, phenotypic differences have been reported in patients with the same variants, even within families. The proband, a girl, was suspected to have in utero fractures of the long bones, suggestive of osteogenesis imperfecta. No respiratory impairment was observed after birth; however, the patient's serum alkaline phosphatase level was low. In addition, the patient's perinatal findings were consistent with those of perinatal benign HPP, although the bone symptoms subsequently worsened. The patient's brother, initially suspected to have odonto-HPP due to the premature loss of primary teeth, later developed compression fractures and extraosseous symptoms. Both patients had the same <i>ALPL</i> variants, c. 572A>G(;)1559del, p. Glu191Gly(;)Leu520ArgfsTer86; however, the severity of their conditions differed. Patients with HPP with identical genotypes in the same family may have varying severity levels of HPP. In this case report, both patients received enzyme replacement therapy (ERT), which improved the clinical symptoms. Therefore, for perinatal benign HPP, ERT should be considered if bone symptoms worsen. In addition, odonto-HPP should be closely monitored, and ERT should be considered if bone and extraosseous symptoms arise.</p>\",\"PeriodicalId\":10678,\"journal\":{\"name\":\"Clinical Pediatric Endocrinology\",\"volume\":\"34 2\",\"pages\":\"137-143\"},\"PeriodicalIF\":1.2000,\"publicationDate\":\"2025-04-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11972865/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Pediatric Endocrinology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1297/cpe.2024-0084\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/2/13 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2024-0084","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/13 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
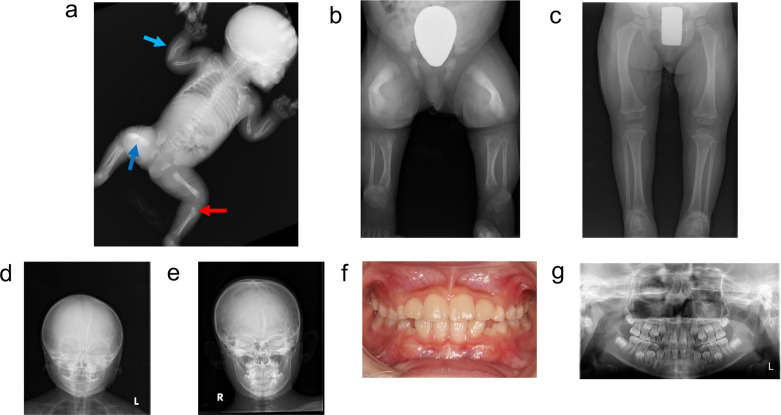
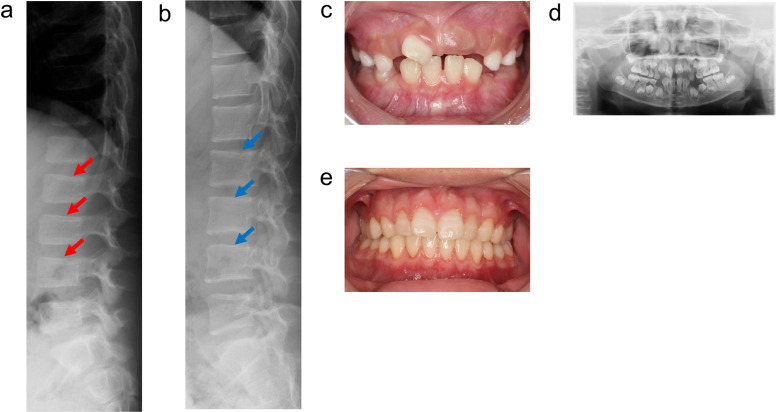
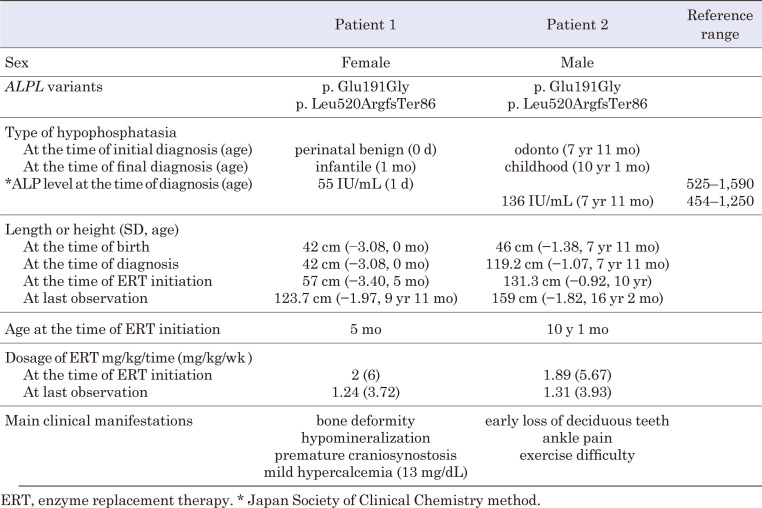
低磷酸症(HPP)是一种遗传性疾病,其特征是由组织非特异性碱性磷酸酶(TNSALP)活性降低引起的骨矿化受损。具体来说,HPP是由编码TNSALP的ALPL基因的功能丧失变体引起的。尽管基因型-表型相关性已被描述,但表型差异已被报道在具有相同变异的患者中,甚至在家庭内。先证者,一名女孩,被怀疑在子宫内有长骨骨折,暗示成骨不完全。出生后未见呼吸障碍;但患者血清碱性磷酸酶水平较低。此外,患者的围产期表现与围产期良性HPP一致,尽管随后骨骼症状恶化。患者的兄弟最初因乳牙过早脱落而怀疑患有牙髓- hpp,后来出现压缩性骨折和骨外症状。两例患者均有相同的ALPL变异,c. 572A>G(;)1559del, p. Glu191Gly(;)Leu520ArgfsTer86;然而,他们的病情严重程度有所不同。同一家族中具有相同基因型的HPP患者可能具有不同的HPP严重程度。在本病例报告中,两名患者均接受了酶替代治疗(ERT),改善了临床症状。因此,对于围产期良性HPP,如果骨骼症状加重,应考虑ERT。此外,应密切监测牙髓- hpp,如果出现骨和骨外症状,应考虑ERT。
Effects of enzyme replacement therapy in sibling cases of hypophosphatasia of varying severities.
Hypophosphatasia (HPP) is a hereditary disorder characterized by impaired bone mineralization caused by decreased tissue-nonspecific alkaline phosphatase (TNSALP) activity. Specifically, HPP is caused by a loss-of-function variant in the ALPL gene encoding TNSALP. Although genotype-phenotype correlations have been described, phenotypic differences have been reported in patients with the same variants, even within families. The proband, a girl, was suspected to have in utero fractures of the long bones, suggestive of osteogenesis imperfecta. No respiratory impairment was observed after birth; however, the patient's serum alkaline phosphatase level was low. In addition, the patient's perinatal findings were consistent with those of perinatal benign HPP, although the bone symptoms subsequently worsened. The patient's brother, initially suspected to have odonto-HPP due to the premature loss of primary teeth, later developed compression fractures and extraosseous symptoms. Both patients had the same ALPL variants, c. 572A>G(;)1559del, p. Glu191Gly(;)Leu520ArgfsTer86; however, the severity of their conditions differed. Patients with HPP with identical genotypes in the same family may have varying severity levels of HPP. In this case report, both patients received enzyme replacement therapy (ERT), which improved the clinical symptoms. Therefore, for perinatal benign HPP, ERT should be considered if bone symptoms worsen. In addition, odonto-HPP should be closely monitored, and ERT should be considered if bone and extraosseous symptoms arise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


