{"title":"解密临床试验时代的面阔肌营养不良症:我们现在在哪里?","authors":"Francesca Torri, Beatrice Ciurli, Mariaconcetta Rende, Arianna Votta, Emanuele Mocciaro, Frida Karakashi, Matteo Lencioni, Elisabetta Ferraro, Massimiliano Filosto, Davide Gabellini, Gabriele Siciliano, Giulia Ricci","doi":"10.36185/2532-1900-1047","DOIUrl":null,"url":null,"abstract":"<p><strong>Objectives: </strong>Facioscapulohumeral muscular dystrophy (FSHD) is a common genetic disorder characterized by progressive muscle weakness, especially in the face, shoulders, and upper limbs. Despite extensive research, the underlying pathogenesis and clinical variability remain incompletely understood. This review aims to summarize recent advances in FSHD research, focusing on genetic and epigenetic factors and the potential for precision medicine.</p><p><strong>Methods: </strong>A comprehensive review of recent literature was conducted, examining molecular mechanisms such as mutations in the D4Z4 region, DUX4 expression, RNA interference (RNAi) and antisense oligonucleotides (AOs). Clinical variability was analyzed to assess different disease phenotypes. Clinical trials investigating potential treatments, especially those targeting DUX4, were also reviewed.</p><p><strong>Results: </strong>FSHD shows significant clinical variability, with different progression rates across phenotypes. The 4qA allele is linked to more typical forms of the disease, but epigenetic factors, including DNA methylation and miRNA expression, also influence disease severity. Despite progress, the exact molecular mechanisms driving disease expression remain unclear. Clinical trials, such as Losmapimod, show promise in slowing muscle degeneration, though results remain inconsistent.</p><p><strong>Conclusions: </strong>FSHD presents significant challenges for therapy development due to its genetic complexity and clinical variability. Ongoing research is needed to clarify pathogenesis and identify reliable biomarkers. Future therapeutic strategies should focus on precision medicine, integrating genetic, clinical, and imaging data to optimize patient stratification and treatment efficacy.</p>","PeriodicalId":93851,"journal":{"name":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","volume":"44 1","pages":"2-10"},"PeriodicalIF":0.0000,"publicationDate":"2025-03-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11978427/pdf/","citationCount":"0","resultStr":"{\"title\":\"Deciphering Facioscapulohumeral Dystrophy in the clinical trials era: where are we now?\",\"authors\":\"Francesca Torri, Beatrice Ciurli, Mariaconcetta Rende, Arianna Votta, Emanuele Mocciaro, Frida Karakashi, Matteo Lencioni, Elisabetta Ferraro, Massimiliano Filosto, Davide Gabellini, Gabriele Siciliano, Giulia Ricci\",\"doi\":\"10.36185/2532-1900-1047\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objectives: </strong>Facioscapulohumeral muscular dystrophy (FSHD) is a common genetic disorder characterized by progressive muscle weakness, especially in the face, shoulders, and upper limbs. Despite extensive research, the underlying pathogenesis and clinical variability remain incompletely understood. This review aims to summarize recent advances in FSHD research, focusing on genetic and epigenetic factors and the potential for precision medicine.</p><p><strong>Methods: </strong>A comprehensive review of recent literature was conducted, examining molecular mechanisms such as mutations in the D4Z4 region, DUX4 expression, RNA interference (RNAi) and antisense oligonucleotides (AOs). Clinical variability was analyzed to assess different disease phenotypes. Clinical trials investigating potential treatments, especially those targeting DUX4, were also reviewed.</p><p><strong>Results: </strong>FSHD shows significant clinical variability, with different progression rates across phenotypes. The 4qA allele is linked to more typical forms of the disease, but epigenetic factors, including DNA methylation and miRNA expression, also influence disease severity. Despite progress, the exact molecular mechanisms driving disease expression remain unclear. Clinical trials, such as Losmapimod, show promise in slowing muscle degeneration, though results remain inconsistent.</p><p><strong>Conclusions: </strong>FSHD presents significant challenges for therapy development due to its genetic complexity and clinical variability. Ongoing research is needed to clarify pathogenesis and identify reliable biomarkers. Future therapeutic strategies should focus on precision medicine, integrating genetic, clinical, and imaging data to optimize patient stratification and treatment efficacy.</p>\",\"PeriodicalId\":93851,\"journal\":{\"name\":\"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology\",\"volume\":\"44 1\",\"pages\":\"2-10\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-03-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11978427/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.36185/2532-1900-1047\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta myologica : myopathies and cardiomyopathies : official journal of the Mediterranean Society of Myology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.36185/2532-1900-1047","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
Deciphering Facioscapulohumeral Dystrophy in the clinical trials era: where are we now?
Objectives: Facioscapulohumeral muscular dystrophy (FSHD) is a common genetic disorder characterized by progressive muscle weakness, especially in the face, shoulders, and upper limbs. Despite extensive research, the underlying pathogenesis and clinical variability remain incompletely understood. This review aims to summarize recent advances in FSHD research, focusing on genetic and epigenetic factors and the potential for precision medicine.
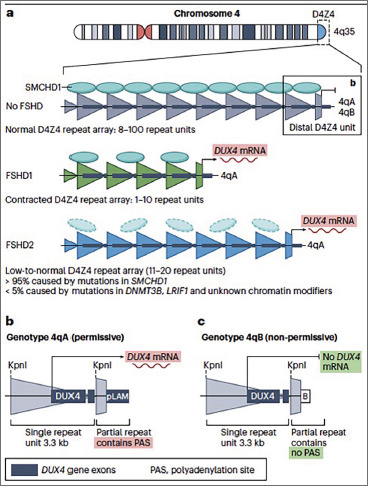
Methods: A comprehensive review of recent literature was conducted, examining molecular mechanisms such as mutations in the D4Z4 region, DUX4 expression, RNA interference (RNAi) and antisense oligonucleotides (AOs). Clinical variability was analyzed to assess different disease phenotypes. Clinical trials investigating potential treatments, especially those targeting DUX4, were also reviewed.
Results: FSHD shows significant clinical variability, with different progression rates across phenotypes. The 4qA allele is linked to more typical forms of the disease, but epigenetic factors, including DNA methylation and miRNA expression, also influence disease severity. Despite progress, the exact molecular mechanisms driving disease expression remain unclear. Clinical trials, such as Losmapimod, show promise in slowing muscle degeneration, though results remain inconsistent.
Conclusions: FSHD presents significant challenges for therapy development due to its genetic complexity and clinical variability. Ongoing research is needed to clarify pathogenesis and identify reliable biomarkers. Future therapeutic strategies should focus on precision medicine, integrating genetic, clinical, and imaging data to optimize patient stratification and treatment efficacy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


