María R Luque-Urbano, David Fernández-Ramos, Fernando Lopitz-Otsoa, Virginia Gutiérrez de Juan, Maider Bizkarguenaga, Lia Castro-Espadas, Uxue Hermoso-Martínez, Lucía Barbier-Torres, Shelly C Lu, Oscar Millet, José M Mato
{"title":"S-Adenosylmethionine 缺乏会破坏极低密度脂蛋白代谢,促进小鼠肝脏脂质积累","authors":"María R Luque-Urbano, David Fernández-Ramos, Fernando Lopitz-Otsoa, Virginia Gutiérrez de Juan, Maider Bizkarguenaga, Lia Castro-Espadas, Uxue Hermoso-Martínez, Lucía Barbier-Torres, Shelly C Lu, Oscar Millet, José M Mato","doi":"10.1016/j.jlr.2025.100794","DOIUrl":null,"url":null,"abstract":"<p><p>Hepatic deletion of methionine adenosyltransferase-1a (Mat1a) in mice reduces S-adenosylmethionine (SAMe), a key methyl donor essential for many biological processes, which promotes the development and progression of metabolic dysfunction-associated steatotic liver disease (MASLD). Hyperglycemia and reduced MAT1A expression, along with low SAMe levels, are common in MASLD patients. This study explores how Mat1a-knockout (KO) hepatocytes respond to prolonged high glucose conditions, focusing on glucose metabolism and lipid accumulation. Hepatocytes from methionine adenosyltransferase-1a-knockout (Mat1a-KO) mice were incubated in high glucose conditions overnight, allowing for analysis of key metabolic intermediates and gene expression related to glycolysis, gluconeogenesis, glyceroneogenesis, phospholipid synthesis, and very low density lipoprotein (VLDL) secretion. SAMe deficiency in Mat1a-KO hepatocytes led to reduced protein methyltransferase-1 activity, resulting in increased expression of glycolytic enzymes (glucokinase, phosphofructokinase, and pyruvate kinase) and decreased expression of gluconeogenic enzymes (phosphoenolpyruvate carboxykinase, fructose-1,6-bisphosphatase, and glucose-6-phosphatase). These alterations led to a reduction in dihydroxyacetone phosphate (DHAP), which subsequently inhibited mammalian target of rapamycin complex 1 (mTORC1) activity. This inhibition resulted in decreased phosphatidylcholine synthesis via the CDP-choline pathway and impaired VLDL secretion, ultimately causing lipid accumulation. Thus, under high glucose conditions, SAMe deficiency in hepatocytes depletes DHAP, inhibits mTORC1 activity, and promotes lipid buildup.</p>","PeriodicalId":16209,"journal":{"name":"Journal of Lipid Research","volume":" ","pages":"100794"},"PeriodicalIF":4.1000,"publicationDate":"2025-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12147229/pdf/","citationCount":"0","resultStr":"{\"title\":\"S-adenosylmethionine deficit disrupts very low-density lipoprotein metabolism promoting liver lipid accumulation in mice.\",\"authors\":\"María R Luque-Urbano, David Fernández-Ramos, Fernando Lopitz-Otsoa, Virginia Gutiérrez de Juan, Maider Bizkarguenaga, Lia Castro-Espadas, Uxue Hermoso-Martínez, Lucía Barbier-Torres, Shelly C Lu, Oscar Millet, José M Mato\",\"doi\":\"10.1016/j.jlr.2025.100794\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Hepatic deletion of methionine adenosyltransferase-1a (Mat1a) in mice reduces S-adenosylmethionine (SAMe), a key methyl donor essential for many biological processes, which promotes the development and progression of metabolic dysfunction-associated steatotic liver disease (MASLD). Hyperglycemia and reduced MAT1A expression, along with low SAMe levels, are common in MASLD patients. This study explores how Mat1a-knockout (KO) hepatocytes respond to prolonged high glucose conditions, focusing on glucose metabolism and lipid accumulation. Hepatocytes from methionine adenosyltransferase-1a-knockout (Mat1a-KO) mice were incubated in high glucose conditions overnight, allowing for analysis of key metabolic intermediates and gene expression related to glycolysis, gluconeogenesis, glyceroneogenesis, phospholipid synthesis, and very low density lipoprotein (VLDL) secretion. SAMe deficiency in Mat1a-KO hepatocytes led to reduced protein methyltransferase-1 activity, resulting in increased expression of glycolytic enzymes (glucokinase, phosphofructokinase, and pyruvate kinase) and decreased expression of gluconeogenic enzymes (phosphoenolpyruvate carboxykinase, fructose-1,6-bisphosphatase, and glucose-6-phosphatase). These alterations led to a reduction in dihydroxyacetone phosphate (DHAP), which subsequently inhibited mammalian target of rapamycin complex 1 (mTORC1) activity. This inhibition resulted in decreased phosphatidylcholine synthesis via the CDP-choline pathway and impaired VLDL secretion, ultimately causing lipid accumulation. Thus, under high glucose conditions, SAMe deficiency in hepatocytes depletes DHAP, inhibits mTORC1 activity, and promotes lipid buildup.</p>\",\"PeriodicalId\":16209,\"journal\":{\"name\":\"Journal of Lipid Research\",\"volume\":\" \",\"pages\":\"100794\"},\"PeriodicalIF\":4.1000,\"publicationDate\":\"2025-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12147229/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Lipid Research\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1016/j.jlr.2025.100794\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/4/1 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Lipid Research","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.jlr.2025.100794","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
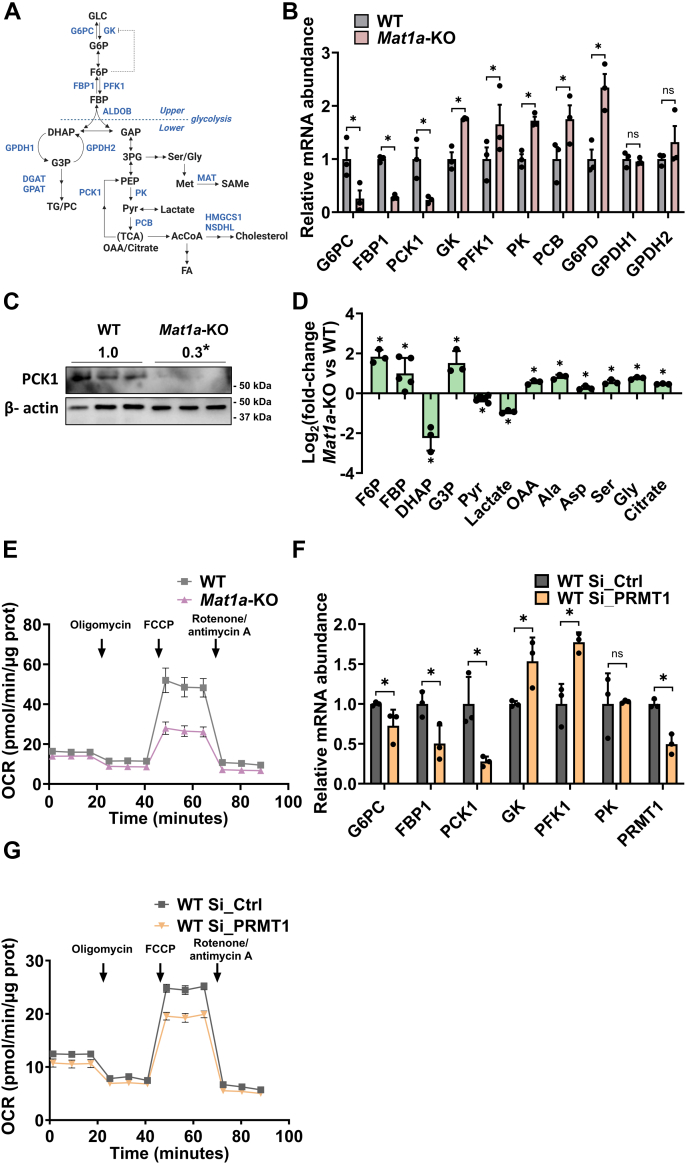
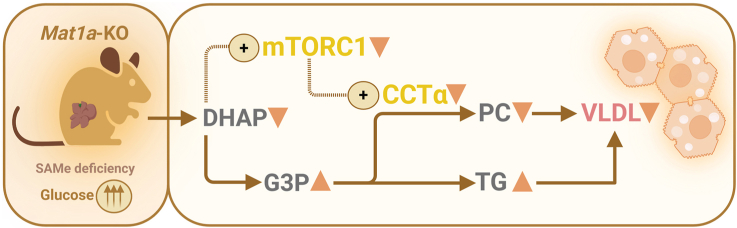
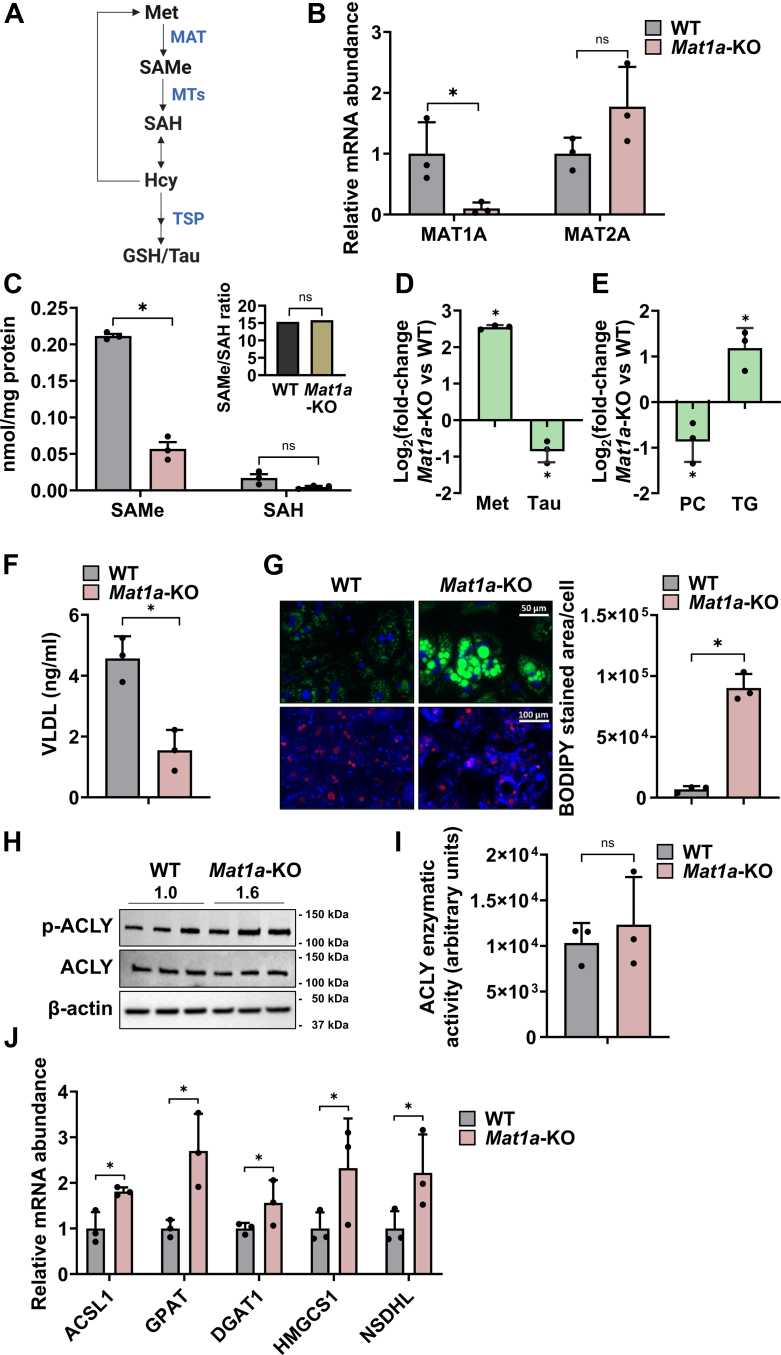
小鼠肝脏中蛋氨酸腺苷转移酶-1a(Mat1a)的缺失会降低S-腺苷蛋氨酸(SAMe),而SAMe是许多生物过程中必不可少的关键甲基供体,它能促进代谢功能障碍相关性脂肪性肝病(MASLD)的发生和发展。高血糖、MAT1A 表达减少以及低 SAMe 水平在 MASLD 患者中很常见。本研究探讨了Mat1a基因敲除(KO)肝细胞如何应对长期高糖条件,重点关注葡萄糖代谢和脂质积累。将 Mat1a-KO 小鼠的肝细胞在高糖条件下培养过夜,可以分析与糖酵解、糖生成、甘油生成、磷脂合成和极低密度脂蛋白(VLDL)分泌有关的关键代谢中间产物和基因表达。Mat1a-KO 肝细胞中缺乏 SAMe 会导致蛋白甲基转移酶-1 活性降低,从而导致糖酵解酶(葡萄糖激酶、磷酸果糖激酶和丙酮酸激酶)表达增加,糖元生成酶(磷酸烯醇丙酮酸羧激酶、果糖-1,6-二磷酸酶和葡萄糖-6-磷酸酶)表达减少。这些变化导致磷酸二羟丙酮(DHAP)减少,进而抑制了哺乳动物雷帕霉素靶复合物 1(mTORC1)的活性。这种抑制导致通过 CDP-choline 途径合成的磷脂酰胆碱减少,VLDL 分泌受阻,最终导致脂质积累。因此,在高糖条件下,肝细胞中缺乏 SAMe 会消耗 DHAP,抑制 mTORC1 的活性,促进脂质堆积。
S-adenosylmethionine deficit disrupts very low-density lipoprotein metabolism promoting liver lipid accumulation in mice.
Hepatic deletion of methionine adenosyltransferase-1a (Mat1a) in mice reduces S-adenosylmethionine (SAMe), a key methyl donor essential for many biological processes, which promotes the development and progression of metabolic dysfunction-associated steatotic liver disease (MASLD). Hyperglycemia and reduced MAT1A expression, along with low SAMe levels, are common in MASLD patients. This study explores how Mat1a-knockout (KO) hepatocytes respond to prolonged high glucose conditions, focusing on glucose metabolism and lipid accumulation. Hepatocytes from methionine adenosyltransferase-1a-knockout (Mat1a-KO) mice were incubated in high glucose conditions overnight, allowing for analysis of key metabolic intermediates and gene expression related to glycolysis, gluconeogenesis, glyceroneogenesis, phospholipid synthesis, and very low density lipoprotein (VLDL) secretion. SAMe deficiency in Mat1a-KO hepatocytes led to reduced protein methyltransferase-1 activity, resulting in increased expression of glycolytic enzymes (glucokinase, phosphofructokinase, and pyruvate kinase) and decreased expression of gluconeogenic enzymes (phosphoenolpyruvate carboxykinase, fructose-1,6-bisphosphatase, and glucose-6-phosphatase). These alterations led to a reduction in dihydroxyacetone phosphate (DHAP), which subsequently inhibited mammalian target of rapamycin complex 1 (mTORC1) activity. This inhibition resulted in decreased phosphatidylcholine synthesis via the CDP-choline pathway and impaired VLDL secretion, ultimately causing lipid accumulation. Thus, under high glucose conditions, SAMe deficiency in hepatocytes depletes DHAP, inhibits mTORC1 activity, and promotes lipid buildup.
期刊介绍:
The Journal of Lipid Research (JLR) publishes original articles and reviews in the broadly defined area of biological lipids. We encourage the submission of manuscripts relating to lipids, including those addressing problems in biochemistry, molecular biology, structural biology, cell biology, genetics, molecular medicine, clinical medicine and metabolism. Major criteria for acceptance of articles are new insights into mechanisms of lipid function and metabolism and/or genes regulating lipid metabolism along with sound primary experimental data. Interpretation of the data is the authors’ responsibility, and speculation should be labeled as such. Manuscripts that provide new ways of purifying, identifying and quantifying lipids are invited for the Methods section of the Journal. JLR encourages contributions from investigators in all countries, but articles must be submitted in clear and concise English.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


