Leandro Benatto, Marlus Koehler, Rodrigo B. Capaz and Graziâni Candiotto
{"title":"用苯丙烯衍生物构件对非富勒烯核心进行改性,实现近乎零的单三元间隙","authors":"Leandro Benatto, Marlus Koehler, Rodrigo B. Capaz and Graziâni Candiotto","doi":"10.1039/D5CP00767D","DOIUrl":null,"url":null,"abstract":"<p >Recent advances in data-driven machine learning have highlighted the critical importance of the singlet–triplet gap (Δ<em>E</em><small><sub>ST</sub></small> = <em>E</em><small><sub>S<small><sub>1</sub></small></sub></small> − <em>E</em><small><sub>T<small><sub>1</sub></small></sub></small>) in non-fullerene acceptor (NFA) molecules as a useful figure of merit to predict the efficiency of organic photovoltaic devices. By reducing Δ<em>E</em><small><sub>ST</sub></small>, the photovoltaic performance can be improved through the suppression of triplet state channels for non-geminate charge recombination. Encouraged by this strategy, we propose and theoretically explore the properties (particularly relative to Δ<em>E</em><small><sub>ST</sub></small>) of a new class of NFAs derived from modifications of the central core of the Y6 molecule (C<small><sub>82</sub></small>H<small><sub>86</sub></small>F<small><sub>4</sub></small>N<small><sub>8</sub></small>O<small><sub>2</sub></small>S<small><sub>5</sub></small>). The idea is to replace the benzothiadiazole chemical group by building blocks of phenalene derivatives, recognized for their unique inverted Δ<em>E</em><small><sub>ST</sub></small>. Using computational analysis that incorporates a double-hybrid exchange–correlation functional as a benchmark method, we anticipate a remarkable reduction of Δ<em>E</em><small><sub>ST</sub></small> upon phenalene derivative substitution, with some molecules achieving a near zero singlet–triplet gap. This is the first report that calls attention to new chemical strategies to synthesize NFA molecules with very low (eventually zero) Δ<em>E</em><small><sub>ST</sub></small>. Moreover, some modified molecules exhibited higher <em>E</em><small><sub>T<small><sub>1</sub></small></sub></small> compared to Y6, which is interesting to mitigate non-geminate recombination. The molecular modifications also lead to a decrease in intramolecular reorganization energy, thereby lowering the energy barrier for electron transfer. Additionally, a significant increase in the quadrupole moment component along the π–π stacking direction was observed—an essential property for strengthening quadrupole–quadrupole intermolecular interactions, which play a crucial role in molecular packing and charge transport. Overall, our research yields valuable insights into optimizing NFAs, opening the possibility of alternative molecular architectures to further the development of high-efficiency organic photovoltaic devices.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 17","pages":" 9112-9122"},"PeriodicalIF":2.9000,"publicationDate":"2025-04-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Near zero singlet–triplet gap through nonfullerene core modification with phenalene derivative building blocks†‡\",\"authors\":\"Leandro Benatto, Marlus Koehler, Rodrigo B. Capaz and Graziâni Candiotto\",\"doi\":\"10.1039/D5CP00767D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Recent advances in data-driven machine learning have highlighted the critical importance of the singlet–triplet gap (Δ<em>E</em><small><sub>ST</sub></small> = <em>E</em><small><sub>S<small><sub>1</sub></small></sub></small> − <em>E</em><small><sub>T<small><sub>1</sub></small></sub></small>) in non-fullerene acceptor (NFA) molecules as a useful figure of merit to predict the efficiency of organic photovoltaic devices. By reducing Δ<em>E</em><small><sub>ST</sub></small>, the photovoltaic performance can be improved through the suppression of triplet state channels for non-geminate charge recombination. Encouraged by this strategy, we propose and theoretically explore the properties (particularly relative to Δ<em>E</em><small><sub>ST</sub></small>) of a new class of NFAs derived from modifications of the central core of the Y6 molecule (C<small><sub>82</sub></small>H<small><sub>86</sub></small>F<small><sub>4</sub></small>N<small><sub>8</sub></small>O<small><sub>2</sub></small>S<small><sub>5</sub></small>). The idea is to replace the benzothiadiazole chemical group by building blocks of phenalene derivatives, recognized for their unique inverted Δ<em>E</em><small><sub>ST</sub></small>. Using computational analysis that incorporates a double-hybrid exchange–correlation functional as a benchmark method, we anticipate a remarkable reduction of Δ<em>E</em><small><sub>ST</sub></small> upon phenalene derivative substitution, with some molecules achieving a near zero singlet–triplet gap. This is the first report that calls attention to new chemical strategies to synthesize NFA molecules with very low (eventually zero) Δ<em>E</em><small><sub>ST</sub></small>. Moreover, some modified molecules exhibited higher <em>E</em><small><sub>T<small><sub>1</sub></small></sub></small> compared to Y6, which is interesting to mitigate non-geminate recombination. The molecular modifications also lead to a decrease in intramolecular reorganization energy, thereby lowering the energy barrier for electron transfer. Additionally, a significant increase in the quadrupole moment component along the π–π stacking direction was observed—an essential property for strengthening quadrupole–quadrupole intermolecular interactions, which play a crucial role in molecular packing and charge transport. Overall, our research yields valuable insights into optimizing NFAs, opening the possibility of alternative molecular architectures to further the development of high-efficiency organic photovoltaic devices.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 17\",\"pages\":\" 9112-9122\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-04-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00767d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00767d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
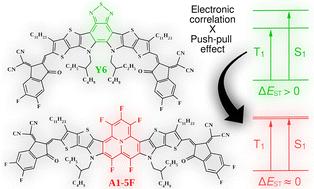
Near zero singlet–triplet gap through nonfullerene core modification with phenalene derivative building blocks†‡
Recent advances in data-driven machine learning have highlighted the critical importance of the singlet–triplet gap (ΔEST = ES1 − ET1) in non-fullerene acceptor (NFA) molecules as a useful figure of merit to predict the efficiency of organic photovoltaic devices. By reducing ΔEST, the photovoltaic performance can be improved through the suppression of triplet state channels for non-geminate charge recombination. Encouraged by this strategy, we propose and theoretically explore the properties (particularly relative to ΔEST) of a new class of NFAs derived from modifications of the central core of the Y6 molecule (C82H86F4N8O2S5). The idea is to replace the benzothiadiazole chemical group by building blocks of phenalene derivatives, recognized for their unique inverted ΔEST. Using computational analysis that incorporates a double-hybrid exchange–correlation functional as a benchmark method, we anticipate a remarkable reduction of ΔEST upon phenalene derivative substitution, with some molecules achieving a near zero singlet–triplet gap. This is the first report that calls attention to new chemical strategies to synthesize NFA molecules with very low (eventually zero) ΔEST. Moreover, some modified molecules exhibited higher ET1 compared to Y6, which is interesting to mitigate non-geminate recombination. The molecular modifications also lead to a decrease in intramolecular reorganization energy, thereby lowering the energy barrier for electron transfer. Additionally, a significant increase in the quadrupole moment component along the π–π stacking direction was observed—an essential property for strengthening quadrupole–quadrupole intermolecular interactions, which play a crucial role in molecular packing and charge transport. Overall, our research yields valuable insights into optimizing NFAs, opening the possibility of alternative molecular architectures to further the development of high-efficiency organic photovoltaic devices.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


