液体电解质开发的预测机器学习力场框架
IF 23.9
1区 计算机科学
Q1 COMPUTER SCIENCE, ARTIFICIAL INTELLIGENCE
引用次数: 0
摘要
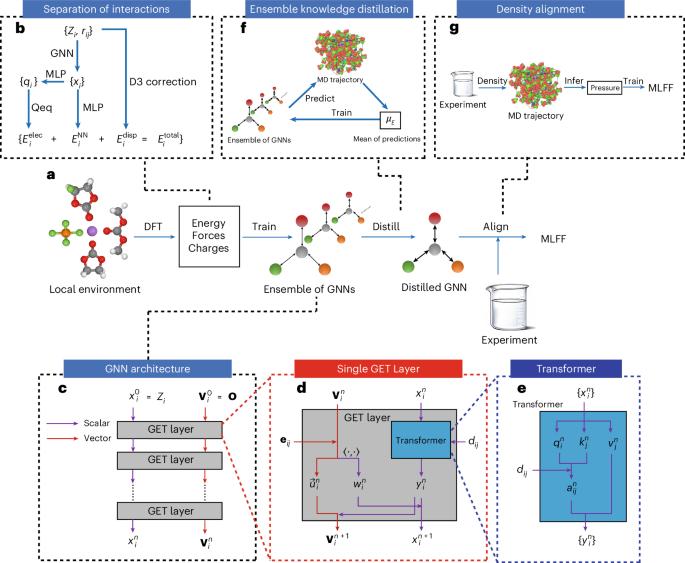
尽管机器学习力场(MLFFs)在固体和小分子中的广泛应用,但在将MLFFs应用于模拟液体电解质(当前商用锂离子电池的关键组成部分)方面存在显着差距。本文介绍了字节跳动人工智能分子模拟助推器(BAMBOO),这是一种用于分子动力学模拟的预测框架,并展示了其在锂电池液体电解质环境中的能力。我们设计了一个物理启发的图形等变变压器架构作为BAMBOO的主干,以学习量子力学模拟。此外,我们引入了一种集成知识蒸馏方法,并将其应用于MLFFs中,以减少分子动力学模拟中观测值的波动。最后,我们提出了一种密度对齐算法,将BAMBOO与实验测量值对齐。BAMBOO在预测各种溶剂和盐组合的关键电解质特性(如密度、粘度和离子电导率)方面展示了最先进的准确性。目前的模型对超过15种化学物质进行了训练,与实验相比,不同成分的平均密度误差为0.01 g cm−3。本文章由计算机程序翻译,如有差异,请以英文原文为准。

A predictive machine learning force-field framework for liquid electrolyte development
Despite the widespread applications of machine learning force fields (MLFFs) in solids and small molecules, there is a notable gap in applying MLFFs to simulate liquid electrolytes—a critical component of current commercial lithium-ion batteries. Here we introduce ByteDance Artificial intelligence Molecular simulation Booster (BAMBOO), a predictive framework for molecular dynamics simulations, with a demonstration of its capability in the context of liquid electrolytes for lithium batteries. We design a physics-inspired graph equivariant transformer architecture as the backbone of BAMBOO to learn from quantum mechanical simulations. Additionally, we introduce an ensemble knowledge distillation approach and apply it to MLFFs to reduce the fluctuation of observations from molecular dynamics simulations. Finally, we propose a density alignment algorithm to align BAMBOO with experimental measurements. BAMBOO demonstrates state-of-the-art accuracy in predicting key electrolyte properties such as density, viscosity and ionic conductivity across various solvents and salt combinations. The current model, trained on more than 15 chemical species, achieves an average density error of 0.01 g cm−3 on various compositions compared with experiment. A machine learning force-field framework is proposed to predict the density, viscosity and ionic conductivity of liquid electrolytes with accuracy that is higher than classical force fields.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature Machine Intelligence
Multiple-
CiteScore
36.90
自引率
2.10%
发文量
127
期刊介绍:
Nature Machine Intelligence is a distinguished publication that presents original research and reviews on various topics in machine learning, robotics, and AI. Our focus extends beyond these fields, exploring their profound impact on other scientific disciplines, as well as societal and industrial aspects. We recognize limitless possibilities wherein machine intelligence can augment human capabilities and knowledge in domains like scientific exploration, healthcare, medical diagnostics, and the creation of safe and sustainable cities, transportation, and agriculture. Simultaneously, we acknowledge the emergence of ethical, social, and legal concerns due to the rapid pace of advancements.
To foster interdisciplinary discussions on these far-reaching implications, Nature Machine Intelligence serves as a platform for dialogue facilitated through Comments, News Features, News & Views articles, and Correspondence. Our goal is to encourage a comprehensive examination of these subjects.
Similar to all Nature-branded journals, Nature Machine Intelligence operates under the guidance of a team of skilled editors. We adhere to a fair and rigorous peer-review process, ensuring high standards of copy-editing and production, swift publication, and editorial independence.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


