Fanjie Xu, Wentao Guo, Feng Wang, Lin Yao, Hongshuai Wang, Fujie Tang, Zhifeng Gao, Linfeng Zhang, Weinan E, Zhong-Qun Tian, Jun Cheng
{"title":"为基于深度学习的核磁共振化学位移预测建立统一的基准和框架。","authors":"Fanjie Xu, Wentao Guo, Feng Wang, Lin Yao, Hongshuai Wang, Fujie Tang, Zhifeng Gao, Linfeng Zhang, Weinan E, Zhong-Qun Tian, Jun Cheng","doi":"10.1038/s43588-025-00783-z","DOIUrl":null,"url":null,"abstract":"The study of structure–spectrum relationships is essential for spectral interpretation, impacting structural elucidation and material design. Predicting spectra from molecular structures is challenging due to their complex relationships. Here we introduce NMRNet, a deep learning framework using the SE(3) Transformer for atomic environment modeling, following a pretraining and fine-tuning paradigm. To support the evaluation of nuclear magnetic resonance chemical shift prediction models, we have established a comprehensive benchmark based on previous research and databases, covering diverse chemical systems. Applying NMRNet to these benchmark datasets, we achieve competitive performance in both liquid-state and solid-state nuclear magnetic resonance datasets, demonstrating its robustness and practical utility in real-world scenarios. Our work helps to advance deep learning applications in analytical and structural chemistry. A deep learning framework (NMRNet) is developed to model atomic environments for predicting NMR chemical shifts. A benchmark dataset, nmrshiftdb2-2024, is also established to provide a standardized source for evaluating NMR models.","PeriodicalId":74246,"journal":{"name":"Nature computational science","volume":"5 4","pages":"292-300"},"PeriodicalIF":18.3000,"publicationDate":"2025-03-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Toward a unified benchmark and framework for deep learning-based prediction of nuclear magnetic resonance chemical shifts\",\"authors\":\"Fanjie Xu, Wentao Guo, Feng Wang, Lin Yao, Hongshuai Wang, Fujie Tang, Zhifeng Gao, Linfeng Zhang, Weinan E, Zhong-Qun Tian, Jun Cheng\",\"doi\":\"10.1038/s43588-025-00783-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The study of structure–spectrum relationships is essential for spectral interpretation, impacting structural elucidation and material design. Predicting spectra from molecular structures is challenging due to their complex relationships. Here we introduce NMRNet, a deep learning framework using the SE(3) Transformer for atomic environment modeling, following a pretraining and fine-tuning paradigm. To support the evaluation of nuclear magnetic resonance chemical shift prediction models, we have established a comprehensive benchmark based on previous research and databases, covering diverse chemical systems. Applying NMRNet to these benchmark datasets, we achieve competitive performance in both liquid-state and solid-state nuclear magnetic resonance datasets, demonstrating its robustness and practical utility in real-world scenarios. Our work helps to advance deep learning applications in analytical and structural chemistry. A deep learning framework (NMRNet) is developed to model atomic environments for predicting NMR chemical shifts. A benchmark dataset, nmrshiftdb2-2024, is also established to provide a standardized source for evaluating NMR models.\",\"PeriodicalId\":74246,\"journal\":{\"name\":\"Nature computational science\",\"volume\":\"5 4\",\"pages\":\"292-300\"},\"PeriodicalIF\":18.3000,\"publicationDate\":\"2025-03-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature computational science\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s43588-025-00783-z\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature computational science","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43588-025-00783-z","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
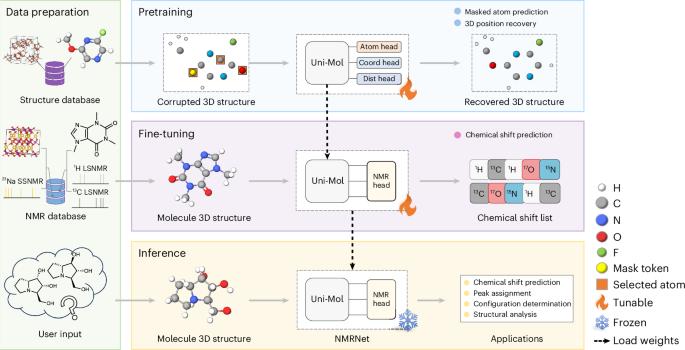
Toward a unified benchmark and framework for deep learning-based prediction of nuclear magnetic resonance chemical shifts
The study of structure–spectrum relationships is essential for spectral interpretation, impacting structural elucidation and material design. Predicting spectra from molecular structures is challenging due to their complex relationships. Here we introduce NMRNet, a deep learning framework using the SE(3) Transformer for atomic environment modeling, following a pretraining and fine-tuning paradigm. To support the evaluation of nuclear magnetic resonance chemical shift prediction models, we have established a comprehensive benchmark based on previous research and databases, covering diverse chemical systems. Applying NMRNet to these benchmark datasets, we achieve competitive performance in both liquid-state and solid-state nuclear magnetic resonance datasets, demonstrating its robustness and practical utility in real-world scenarios. Our work helps to advance deep learning applications in analytical and structural chemistry. A deep learning framework (NMRNet) is developed to model atomic environments for predicting NMR chemical shifts. A benchmark dataset, nmrshiftdb2-2024, is also established to provide a standardized source for evaluating NMR models.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


