Georg F Vogel, Katharina M C Klee, Arzu Meltem Demir, Dorota Garczarczyk-Asim, Michael W Hess, Lukas A Huber, Thomas Müller, Andreas R Janecke
{"title":"由ezrin缺乏引起的先天性肠病。","authors":"Georg F Vogel, Katharina M C Klee, Arzu Meltem Demir, Dorota Garczarczyk-Asim, Michael W Hess, Lukas A Huber, Thomas Müller, Andreas R Janecke","doi":"10.1007/s00439-025-02738-w","DOIUrl":null,"url":null,"abstract":"<p><p>Ezrin, encoded by EZR, is a central module of epithelial polarity and links membrane proteins to the actin cytoskeleton directly or indirectly through scaffold proteins in the epithelium. Ezrin knockout mice fail to thrive and do not survive past weaning. We identified a homozygous EZR loss-of-function (LoF) variant, c.356dup, by exome sequencing in an infant with intractable diarrhea and failure to thrive, who died from septicemia at 5 months of age. The variant localized within a homozygous region of 13.2 Mb in the proband, is consistent with inheritance identical-by-descent from the consanguineous parents, and segregated with disease in the proband's family. EZR transcript analyses in a heterozygous carrier showed that the variant triggers nonsense-mediated mRNA decay. Homozygous EZR LoF variants have not been reported in public databases. In this study, we generated a Caco-2 EZR knockout cell line to investigate the role of ezrin in human intestinal epithelia. Our analyses used electron and immunofluorescence microscopy to assess structural changes in the knockout cells. We observed significant disorganization of the terminal web region, microvillus rarefaction and abnormal branching. Furthermore, the absence of ezrin resulted in the mislocalization of the ezrin-interacting scaffold protein Na+/H + exchanger regulatory factor-1. In conclusion, this represents the first documentation of complete ezrin deficiency in humans, highlighting the essential and non-redundant functions of the protein in maintaining intestinal physiology.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"505-514"},"PeriodicalIF":3.6000,"publicationDate":"2025-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12033192/pdf/","citationCount":"0","resultStr":"{\"title\":\"Congenital enteropathy caused by ezrin deficiency.\",\"authors\":\"Georg F Vogel, Katharina M C Klee, Arzu Meltem Demir, Dorota Garczarczyk-Asim, Michael W Hess, Lukas A Huber, Thomas Müller, Andreas R Janecke\",\"doi\":\"10.1007/s00439-025-02738-w\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Ezrin, encoded by EZR, is a central module of epithelial polarity and links membrane proteins to the actin cytoskeleton directly or indirectly through scaffold proteins in the epithelium. Ezrin knockout mice fail to thrive and do not survive past weaning. We identified a homozygous EZR loss-of-function (LoF) variant, c.356dup, by exome sequencing in an infant with intractable diarrhea and failure to thrive, who died from septicemia at 5 months of age. The variant localized within a homozygous region of 13.2 Mb in the proband, is consistent with inheritance identical-by-descent from the consanguineous parents, and segregated with disease in the proband's family. EZR transcript analyses in a heterozygous carrier showed that the variant triggers nonsense-mediated mRNA decay. Homozygous EZR LoF variants have not been reported in public databases. In this study, we generated a Caco-2 EZR knockout cell line to investigate the role of ezrin in human intestinal epithelia. Our analyses used electron and immunofluorescence microscopy to assess structural changes in the knockout cells. We observed significant disorganization of the terminal web region, microvillus rarefaction and abnormal branching. Furthermore, the absence of ezrin resulted in the mislocalization of the ezrin-interacting scaffold protein Na+/H + exchanger regulatory factor-1. In conclusion, this represents the first documentation of complete ezrin deficiency in humans, highlighting the essential and non-redundant functions of the protein in maintaining intestinal physiology.</p>\",\"PeriodicalId\":13175,\"journal\":{\"name\":\"Human Genetics\",\"volume\":\" \",\"pages\":\"505-514\"},\"PeriodicalIF\":3.6000,\"publicationDate\":\"2025-05-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12033192/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human Genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1007/s00439-025-02738-w\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-025-02738-w","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/26 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Congenital enteropathy caused by ezrin deficiency.
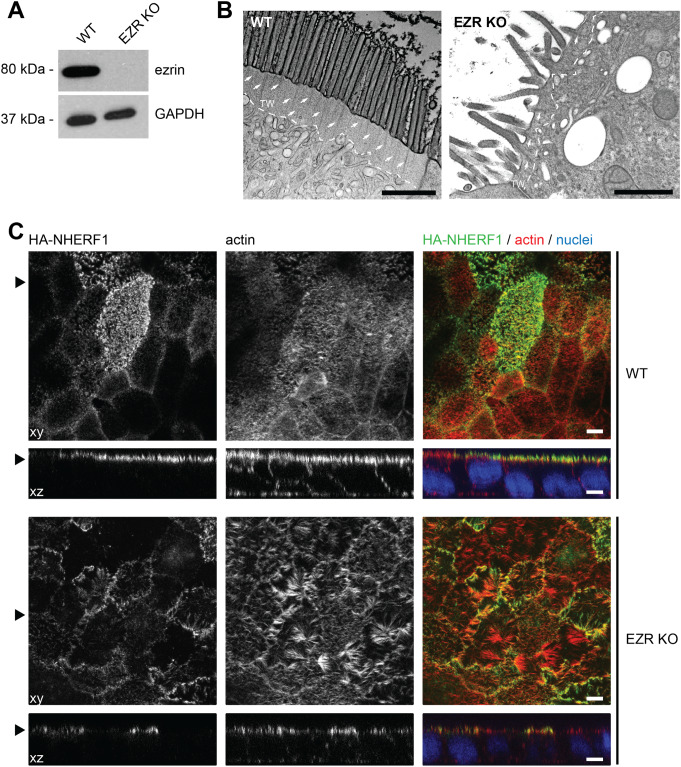
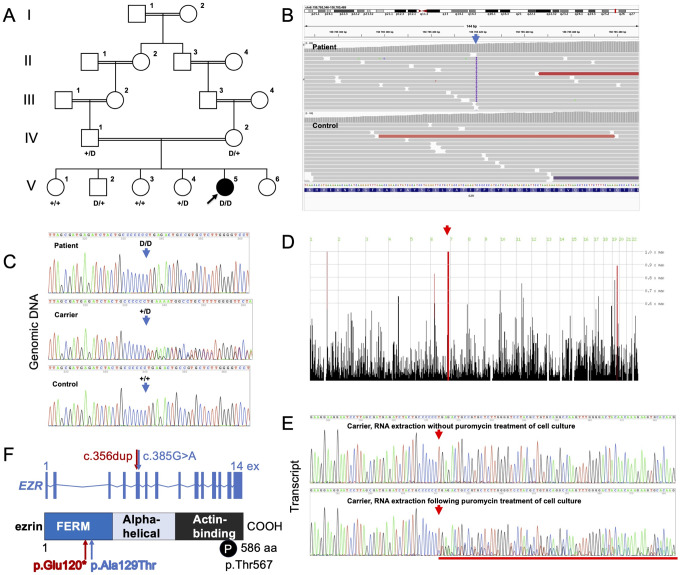
Ezrin, encoded by EZR, is a central module of epithelial polarity and links membrane proteins to the actin cytoskeleton directly or indirectly through scaffold proteins in the epithelium. Ezrin knockout mice fail to thrive and do not survive past weaning. We identified a homozygous EZR loss-of-function (LoF) variant, c.356dup, by exome sequencing in an infant with intractable diarrhea and failure to thrive, who died from septicemia at 5 months of age. The variant localized within a homozygous region of 13.2 Mb in the proband, is consistent with inheritance identical-by-descent from the consanguineous parents, and segregated with disease in the proband's family. EZR transcript analyses in a heterozygous carrier showed that the variant triggers nonsense-mediated mRNA decay. Homozygous EZR LoF variants have not been reported in public databases. In this study, we generated a Caco-2 EZR knockout cell line to investigate the role of ezrin in human intestinal epithelia. Our analyses used electron and immunofluorescence microscopy to assess structural changes in the knockout cells. We observed significant disorganization of the terminal web region, microvillus rarefaction and abnormal branching. Furthermore, the absence of ezrin resulted in the mislocalization of the ezrin-interacting scaffold protein Na+/H + exchanger regulatory factor-1. In conclusion, this represents the first documentation of complete ezrin deficiency in humans, highlighting the essential and non-redundant functions of the protein in maintaining intestinal physiology.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


