{"title":"当一氧化碳“倒过来”:从从头算分子动力学的CO在NaCl(100)中的振动特征","authors":"Shreya Sinha, Alec M. Wodtke and Peter Saalfrank","doi":"10.1039/D4CP04671D","DOIUrl":null,"url":null,"abstract":"<p >CO adsorbed on NaCl(100) is a model system for surface science showing a rich variety of interesting phenomena. It features several adsorption phases like tilted/antiparallel or perpendicular/upright, very long vibrational lifetimes of the CO internal stretch (IS) mode, anharmonicity-driven vibrational energy pooling, “C-bound” <em>vs.</em> “O-bound” adsorption, and heavy-atom gateway tunneling during CO inversion at low temperatures. Typically, these features and phenomena are experimentally probed by stationary and time-resolved vibrational spectra, exhibiting characteristic differences between the various adsorption modes and phases. To gain atom- and time-resolved insight into vibrational response of CO molecules on NaCl(100), vibrational density of states (VDOS), infrared (IR) and vibrational sum frequency (VSF) spectra are computed from velocity velocity correlation functions (VVCFs) by <em>ab initio</em> molecular dynamics (AIMD) for various coverages, temperatures and phases. In agreement with experiments, we find that increasing CO (“C-bound”) coverages as well as CO inversion lead to redshifts of the CO IS mode. We predict more diffuse spectra at <em>T</em> = 300 K compared to 30 K, reflecting the disorder of adsorbates and monolayer instability at room temperature. Analyzing molecularly decomposed and internal VDOS curves as well as computed non-linear correlation matrices give further insight into the complex molecular dynamics underlying the vibrational spectra, notably for the low-frequency regime where frustrated rotations, translations and intermolecular motions come into play. On a methodological side, we also test and discuss some intricate details of how to compute IR and VSF response using a modified formulation of the VVCF methods [Ohto <em>et al.</em>, <em>J. Chem. Phys.</em>, 2015, <strong>143</strong>, 124702], by including time and angle-dependent dipole and polarizability derivatives as well as intermolecular couplings by cross correlations. Their effect on computed vibrational spectra is studied. These findings provide a detailed, microscopic insight into the picosecond vibrational spectra and dynamics of CO on NaCl(100), highlighting the effects of temperature, coverage, and changes in adsorbate orientation.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 15","pages":" 7929-7942"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04671d?page=search","citationCount":"0","resultStr":"{\"title\":\"When carbon monoxide goes “upside down”: vibrational signatures of CO at NaCl(100) from ab initio molecular dynamics†\",\"authors\":\"Shreya Sinha, Alec M. Wodtke and Peter Saalfrank\",\"doi\":\"10.1039/D4CP04671D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >CO adsorbed on NaCl(100) is a model system for surface science showing a rich variety of interesting phenomena. It features several adsorption phases like tilted/antiparallel or perpendicular/upright, very long vibrational lifetimes of the CO internal stretch (IS) mode, anharmonicity-driven vibrational energy pooling, “C-bound” <em>vs.</em> “O-bound” adsorption, and heavy-atom gateway tunneling during CO inversion at low temperatures. Typically, these features and phenomena are experimentally probed by stationary and time-resolved vibrational spectra, exhibiting characteristic differences between the various adsorption modes and phases. To gain atom- and time-resolved insight into vibrational response of CO molecules on NaCl(100), vibrational density of states (VDOS), infrared (IR) and vibrational sum frequency (VSF) spectra are computed from velocity velocity correlation functions (VVCFs) by <em>ab initio</em> molecular dynamics (AIMD) for various coverages, temperatures and phases. In agreement with experiments, we find that increasing CO (“C-bound”) coverages as well as CO inversion lead to redshifts of the CO IS mode. We predict more diffuse spectra at <em>T</em> = 300 K compared to 30 K, reflecting the disorder of adsorbates and monolayer instability at room temperature. Analyzing molecularly decomposed and internal VDOS curves as well as computed non-linear correlation matrices give further insight into the complex molecular dynamics underlying the vibrational spectra, notably for the low-frequency regime where frustrated rotations, translations and intermolecular motions come into play. On a methodological side, we also test and discuss some intricate details of how to compute IR and VSF response using a modified formulation of the VVCF methods [Ohto <em>et al.</em>, <em>J. Chem. Phys.</em>, 2015, <strong>143</strong>, 124702], by including time and angle-dependent dipole and polarizability derivatives as well as intermolecular couplings by cross correlations. Their effect on computed vibrational spectra is studied. These findings provide a detailed, microscopic insight into the picosecond vibrational spectra and dynamics of CO on NaCl(100), highlighting the effects of temperature, coverage, and changes in adsorbate orientation.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 15\",\"pages\":\" 7929-7942\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-03-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04671d?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04671d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04671d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
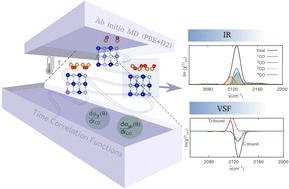
吸附在 NaCl(100)上的一氧化碳是表面科学的一个模型系统,显示出丰富多彩的有趣现象。它具有几种吸附相,如倾斜/反平行或垂直/直立、CO 内部伸展(IS)模式的超长振动寿命、非谐波驱动的振动能量汇集、"C-结合 "与 "O-结合 "吸附,以及低温下 CO 反转过程中的重原子网关隧道。通常情况下,这些特征和现象可通过静态和时间分辨振动光谱进行实验探测,从而显示出各种吸附模式和阶段之间的特征差异。为了从原子和时间分辨的角度深入了解一氧化碳分子在 NaCl(100)上的振动响应,我们通过 Ab Initio 分子动力学(AIMD)计算了不同覆盖率、温度和相位下的速度相关函数(VVCF)的振动状态密度(VDOS)、红外(IR)和振动和频(VSF)光谱。与实验结果一致,我们发现 CO("C-bound")覆盖率的增加以及 CO 的反转会导致 CO IS 模式的红移。我们预测,与 30 K 相比,T = 300 K 时的光谱更分散,这反映了室温下吸附剂的无序性和单层的不稳定性。通过分析分子分解和内部 VDOS 曲线以及计算的非线性相关矩阵,我们进一步了解了振动光谱背后复杂的分子动力学,尤其是在低频体系中,受挫旋转、平移和分子间的摩尔效应都会发挥作用。在方法论方面,我们还测试并讨论了如何使用 VVCF 方法的改进公式计算红外和 VSF 响应的一些复杂细节[Ohto 等人,J. Chem. Phys.,2015,143,124702],方法包括与时间和角度相关的偶极子和极化性导数以及通过交叉相关的分子间耦合。研究了它们对计算振动光谱的影响。这些发现从微观角度详细揭示了一氧化碳在氯化钠(100)上的皮秒振动光谱和动力学,突出了温度、覆盖率和吸附剂取向变化的影响。
When carbon monoxide goes “upside down”: vibrational signatures of CO at NaCl(100) from ab initio molecular dynamics†
CO adsorbed on NaCl(100) is a model system for surface science showing a rich variety of interesting phenomena. It features several adsorption phases like tilted/antiparallel or perpendicular/upright, very long vibrational lifetimes of the CO internal stretch (IS) mode, anharmonicity-driven vibrational energy pooling, “C-bound” vs. “O-bound” adsorption, and heavy-atom gateway tunneling during CO inversion at low temperatures. Typically, these features and phenomena are experimentally probed by stationary and time-resolved vibrational spectra, exhibiting characteristic differences between the various adsorption modes and phases. To gain atom- and time-resolved insight into vibrational response of CO molecules on NaCl(100), vibrational density of states (VDOS), infrared (IR) and vibrational sum frequency (VSF) spectra are computed from velocity velocity correlation functions (VVCFs) by ab initio molecular dynamics (AIMD) for various coverages, temperatures and phases. In agreement with experiments, we find that increasing CO (“C-bound”) coverages as well as CO inversion lead to redshifts of the CO IS mode. We predict more diffuse spectra at T = 300 K compared to 30 K, reflecting the disorder of adsorbates and monolayer instability at room temperature. Analyzing molecularly decomposed and internal VDOS curves as well as computed non-linear correlation matrices give further insight into the complex molecular dynamics underlying the vibrational spectra, notably for the low-frequency regime where frustrated rotations, translations and intermolecular motions come into play. On a methodological side, we also test and discuss some intricate details of how to compute IR and VSF response using a modified formulation of the VVCF methods [Ohto et al., J. Chem. Phys., 2015, 143, 124702], by including time and angle-dependent dipole and polarizability derivatives as well as intermolecular couplings by cross correlations. Their effect on computed vibrational spectra is studied. These findings provide a detailed, microscopic insight into the picosecond vibrational spectra and dynamics of CO on NaCl(100), highlighting the effects of temperature, coverage, and changes in adsorbate orientation.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


