Olga O Lebedenko, Mikhail S Polovinkin, Anastasiia A Kazovskaia, Nikolai R Skrynnikov
{"title":"基于结构的蛋白质结合亲和力预测的PCANN程序:与其他神经网络预测器的比较。","authors":"Olga O Lebedenko, Mikhail S Polovinkin, Anastasiia A Kazovskaia, Nikolai R Skrynnikov","doi":"10.1002/prot.26821","DOIUrl":null,"url":null,"abstract":"<p><p>In this communication, we introduce a new structure-based affinity predictor for protein-protein complexes. This predictor, dubbed PCANN (Protein Complex Affinity by Neural Network), uses the ESM-2 language model to encode the information about protein binding interfaces and graph attention network (GAT) to parlay this information into <math> <semantics> <mrow><msub><mi>K</mi> <mi>d</mi></msub> </mrow> </semantics> </math> predictions. In the tests employing two previously unused literature-extracted datasets, PCANN performed better than the best of the publicly available predictors, BindPPI, with mean absolute error (MAE) of 1.3 versus 1.4 kcal/mol. Further progress in the development of <math> <semantics> <mrow><msub><mi>K</mi> <mi>d</mi></msub> </mrow> </semantics> </math> predictors using deep learning models is faced with two problems: (i) the amount of experimental data available to train and test new predictors is limited and (ii) the available <math> <semantics> <mrow><msub><mi>K</mi> <mi>d</mi></msub> </mrow> </semantics> </math> data are often not very accurate and lack internal consistency with respect to measurement conditions. These issues can be potentially addressed through an AI-leveraged literature search followed by careful human curation and by introducing additional parameters to account for variations in experimental conditions.</p>","PeriodicalId":56271,"journal":{"name":"Proteins-Structure Function and Bioinformatics","volume":" ","pages":"1498-1506"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12314579/pdf/","citationCount":"0","resultStr":"{\"title\":\"PCANN Program for Structure-Based Prediction of Protein-Protein Binding Affinity: Comparison With Other Neural-Network Predictors.\",\"authors\":\"Olga O Lebedenko, Mikhail S Polovinkin, Anastasiia A Kazovskaia, Nikolai R Skrynnikov\",\"doi\":\"10.1002/prot.26821\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>In this communication, we introduce a new structure-based affinity predictor for protein-protein complexes. This predictor, dubbed PCANN (Protein Complex Affinity by Neural Network), uses the ESM-2 language model to encode the information about protein binding interfaces and graph attention network (GAT) to parlay this information into <math> <semantics> <mrow><msub><mi>K</mi> <mi>d</mi></msub> </mrow> </semantics> </math> predictions. In the tests employing two previously unused literature-extracted datasets, PCANN performed better than the best of the publicly available predictors, BindPPI, with mean absolute error (MAE) of 1.3 versus 1.4 kcal/mol. Further progress in the development of <math> <semantics> <mrow><msub><mi>K</mi> <mi>d</mi></msub> </mrow> </semantics> </math> predictors using deep learning models is faced with two problems: (i) the amount of experimental data available to train and test new predictors is limited and (ii) the available <math> <semantics> <mrow><msub><mi>K</mi> <mi>d</mi></msub> </mrow> </semantics> </math> data are often not very accurate and lack internal consistency with respect to measurement conditions. These issues can be potentially addressed through an AI-leveraged literature search followed by careful human curation and by introducing additional parameters to account for variations in experimental conditions.</p>\",\"PeriodicalId\":56271,\"journal\":{\"name\":\"Proteins-Structure Function and Bioinformatics\",\"volume\":\" \",\"pages\":\"1498-1506\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12314579/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Proteins-Structure Function and Bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1002/prot.26821\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteins-Structure Function and Bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1002/prot.26821","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
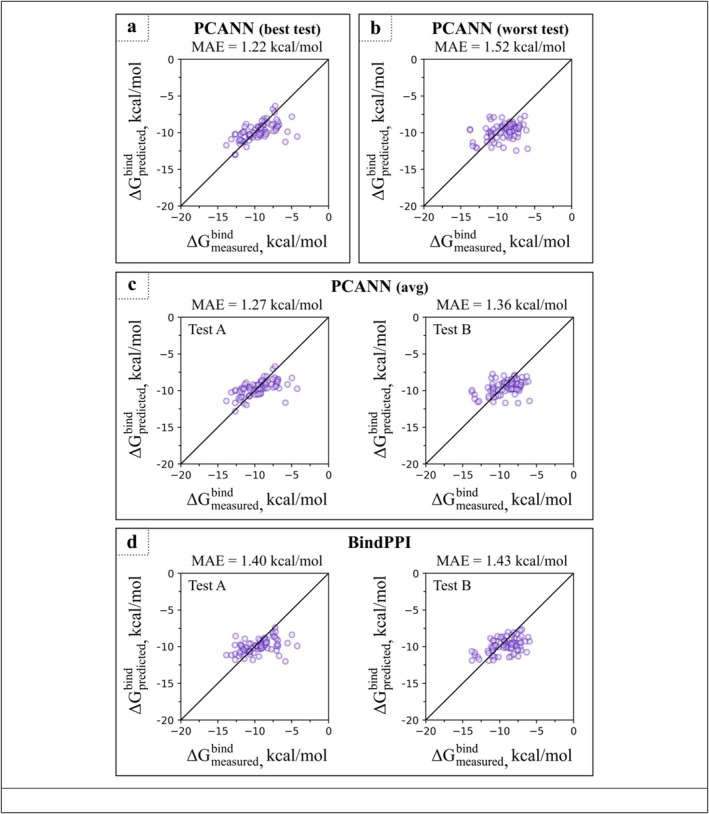
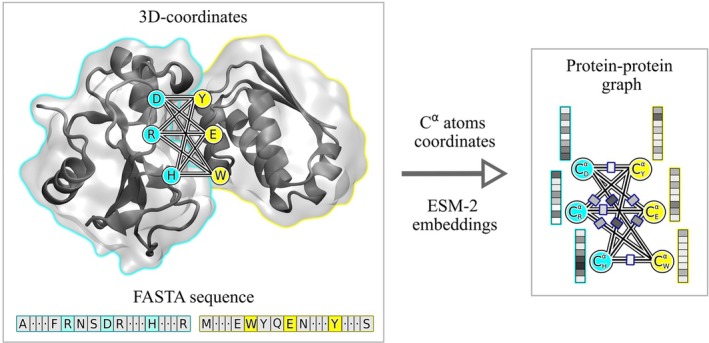
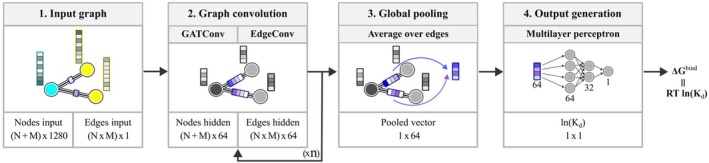
在这篇文章中,我们介绍了一种新的基于结构的蛋白质-蛋白质复合物亲和预测器。这个预测器被称为PCANN(神经网络蛋白质复合物亲和力),它使用ESM-2语言模型对蛋白质结合界面的信息进行编码,并使用图形注意网络(GAT)将这些信息运用到K d $$ {K}_{\mathrm{d}} $$预测中。在使用两个以前未使用的文献提取数据集的测试中,PCANN表现优于公开可用的最佳预测因子BindPPI,平均绝对误差(MAE)为1.3与1.4 kcal/mol。使用深度学习模型开发K d $$ {K}_{\mathrm{d}} $$预测器的进一步进展面临两个问题:(i)可用于训练和测试新预测器的实验数据量有限,(ii)可用的K d $$ {K}_{\mathrm{d}} $$数据通常不是很准确,并且在测量条件方面缺乏内部一致性。这些问题可以通过利用人工智能进行文献搜索,然后进行仔细的人工管理,并引入额外的参数来解释实验条件的变化,从而潜在地解决。
PCANN Program for Structure-Based Prediction of Protein-Protein Binding Affinity: Comparison With Other Neural-Network Predictors.
In this communication, we introduce a new structure-based affinity predictor for protein-protein complexes. This predictor, dubbed PCANN (Protein Complex Affinity by Neural Network), uses the ESM-2 language model to encode the information about protein binding interfaces and graph attention network (GAT) to parlay this information into predictions. In the tests employing two previously unused literature-extracted datasets, PCANN performed better than the best of the publicly available predictors, BindPPI, with mean absolute error (MAE) of 1.3 versus 1.4 kcal/mol. Further progress in the development of predictors using deep learning models is faced with two problems: (i) the amount of experimental data available to train and test new predictors is limited and (ii) the available data are often not very accurate and lack internal consistency with respect to measurement conditions. These issues can be potentially addressed through an AI-leveraged literature search followed by careful human curation and by introducing additional parameters to account for variations in experimental conditions.
期刊介绍:
PROTEINS : Structure, Function, and Bioinformatics publishes original reports of significant experimental and analytic research in all areas of protein research: structure, function, computation, genetics, and design. The journal encourages reports that present new experimental or computational approaches for interpreting and understanding data from biophysical chemistry, structural studies of proteins and macromolecular assemblies, alterations of protein structure and function engineered through techniques of molecular biology and genetics, functional analyses under physiologic conditions, as well as the interactions of proteins with receptors, nucleic acids, or other specific ligands or substrates. Research in protein and peptide biochemistry directed toward synthesizing or characterizing molecules that simulate aspects of the activity of proteins, or that act as inhibitors of protein function, is also within the scope of PROTEINS. In addition to full-length reports, short communications (usually not more than 4 printed pages) and prediction reports are welcome. Reviews are typically by invitation; authors are encouraged to submit proposed topics for consideration.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


