{"title":"多环芳烃在石墨上的稳定性:抗水平位移","authors":"Yoshihiro Kikkawa and Seiji Tsuzuki","doi":"10.1039/D5CP00567A","DOIUrl":null,"url":null,"abstract":"<p >The stability of adsorbed molecules on surfaces depends on the magnitude of adsorbate–substrate interaction energies and the resistance of these molecules to horizontal displacement. Therefore, it is essential to analyse both interaction energy (<em>E</em><small><sub>int</sub></small>) and its change with horizontal displacement (Δ<em>E</em><small><sub>int</sub></small>). In physisorbed monolayers at a highly oriented pyrolytic graphite (HOPG)/solvent interface, molecular building blocks often contain both aromatic and alkyl chain moieties, as aromatic molecules without additional substituents are difficult to observe <em>via</em> scanning tunnelling microscopy. This suggests that aromatic molecules have less adsorption stability on the HOPG than <em>n</em>-alkanes, though the underlying reason remains unclear. In this study, we performed dispersion-corrected density functional theory calculations to evaluate <em>E</em><small><sub>int</sub></small> and Δ<em>E</em><small><sub>int</sub></small> of polycyclic aromatic hydrocarbons (PAHs) on a graphite model surface (C<small><sub>96</sub></small>H<small><sub>24</sub></small>). Δ<em>E</em><small><sub>int</sub></small> was analysed for PAHs with the number of carbon atoms (<em>N</em><small><sub>c</sub></small>) from 6 to 24. The maximum Δ<em>E</em><small><sub>int</sub></small> (Δ<em>E</em><small><sub>int(max)</sub></small>) is related to the barrier height for lateral migration. The Δ<em>E</em><small><sub>int(max)</sub></small> per <em>N</em><small><sub>c</sub></small> showed directional dependence and ranged from 0.015 to 0.20 kcal mol<small><sup>−1</sup></small>, with the largest value for PAHs being about two-thirds that of <em>n</em>-alkanes (0.30 kcal mol<small><sup>−1</sup></small>), indicating greater mobility of the former. These findings demonstrate that aromatic and alkyl chain moieties in two-dimensional assemblies exhibit distinct resistance against horizontal migration. The preferential role of alkyl chains suggests that molecular assemblies align with the graphite lattice, prioritising alkyl unit positioning over aromatic orientation.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 14","pages":" 7421-7428"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Stability of polycyclic aromatic hydrocarbons on graphite: resistance to horizontal displacement†\",\"authors\":\"Yoshihiro Kikkawa and Seiji Tsuzuki\",\"doi\":\"10.1039/D5CP00567A\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The stability of adsorbed molecules on surfaces depends on the magnitude of adsorbate–substrate interaction energies and the resistance of these molecules to horizontal displacement. Therefore, it is essential to analyse both interaction energy (<em>E</em><small><sub>int</sub></small>) and its change with horizontal displacement (Δ<em>E</em><small><sub>int</sub></small>). In physisorbed monolayers at a highly oriented pyrolytic graphite (HOPG)/solvent interface, molecular building blocks often contain both aromatic and alkyl chain moieties, as aromatic molecules without additional substituents are difficult to observe <em>via</em> scanning tunnelling microscopy. This suggests that aromatic molecules have less adsorption stability on the HOPG than <em>n</em>-alkanes, though the underlying reason remains unclear. In this study, we performed dispersion-corrected density functional theory calculations to evaluate <em>E</em><small><sub>int</sub></small> and Δ<em>E</em><small><sub>int</sub></small> of polycyclic aromatic hydrocarbons (PAHs) on a graphite model surface (C<small><sub>96</sub></small>H<small><sub>24</sub></small>). Δ<em>E</em><small><sub>int</sub></small> was analysed for PAHs with the number of carbon atoms (<em>N</em><small><sub>c</sub></small>) from 6 to 24. The maximum Δ<em>E</em><small><sub>int</sub></small> (Δ<em>E</em><small><sub>int(max)</sub></small>) is related to the barrier height for lateral migration. The Δ<em>E</em><small><sub>int(max)</sub></small> per <em>N</em><small><sub>c</sub></small> showed directional dependence and ranged from 0.015 to 0.20 kcal mol<small><sup>−1</sup></small>, with the largest value for PAHs being about two-thirds that of <em>n</em>-alkanes (0.30 kcal mol<small><sup>−1</sup></small>), indicating greater mobility of the former. These findings demonstrate that aromatic and alkyl chain moieties in two-dimensional assemblies exhibit distinct resistance against horizontal migration. The preferential role of alkyl chains suggests that molecular assemblies align with the graphite lattice, prioritising alkyl unit positioning over aromatic orientation.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 14\",\"pages\":\" 7421-7428\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-03-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00567a\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00567a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Stability of polycyclic aromatic hydrocarbons on graphite: resistance to horizontal displacement†
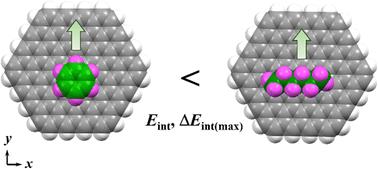
The stability of adsorbed molecules on surfaces depends on the magnitude of adsorbate–substrate interaction energies and the resistance of these molecules to horizontal displacement. Therefore, it is essential to analyse both interaction energy (Eint) and its change with horizontal displacement (ΔEint). In physisorbed monolayers at a highly oriented pyrolytic graphite (HOPG)/solvent interface, molecular building blocks often contain both aromatic and alkyl chain moieties, as aromatic molecules without additional substituents are difficult to observe via scanning tunnelling microscopy. This suggests that aromatic molecules have less adsorption stability on the HOPG than n-alkanes, though the underlying reason remains unclear. In this study, we performed dispersion-corrected density functional theory calculations to evaluate Eint and ΔEint of polycyclic aromatic hydrocarbons (PAHs) on a graphite model surface (C96H24). ΔEint was analysed for PAHs with the number of carbon atoms (Nc) from 6 to 24. The maximum ΔEint (ΔEint(max)) is related to the barrier height for lateral migration. The ΔEint(max) per Nc showed directional dependence and ranged from 0.015 to 0.20 kcal mol−1, with the largest value for PAHs being about two-thirds that of n-alkanes (0.30 kcal mol−1), indicating greater mobility of the former. These findings demonstrate that aromatic and alkyl chain moieties in two-dimensional assemblies exhibit distinct resistance against horizontal migration. The preferential role of alkyl chains suggests that molecular assemblies align with the graphite lattice, prioritising alkyl unit positioning over aromatic orientation.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


