Fatemeh Jamalinabijan, Somayyeh Alidoust, Gözde İniş Demir and Adem Tekin
{"title":"通过机器学习发现新型无铅混合阳离子卤化物钙钛矿","authors":"Fatemeh Jamalinabijan, Somayyeh Alidoust, Gözde İniş Demir and Adem Tekin","doi":"10.1039/D4CP04218B","DOIUrl":null,"url":null,"abstract":"<p >In our recent study (S. Alidoust, F. Jamalinabijan and A. Tekin, <em>ACS Appl. Energy Mater.</em>, 2024, <strong>7</strong>, 785–798), a thorough computational screening using density functional theory (DFT) was conducted on mixed cation halide perovskites with a general formula of AA′BX<small><sub>3</sub></small>, aiming to identify promising lead-free candidates. Employment of 23 A/A′-cations, 29 B-ions, and 4 X-anions yielded approximately 29 000 possible perovskite combinations. However, while modern high-throughput DFT frameworks can handle large-scale calculations, treating the entire configurational space of 29 000 possible perovskite combinations remains computationally demanding. Leveraging machine learning (ML) approaches could provide a more efficient alternative for capturing this complexity. Therefore, by using two empirical criteria known as octahedral and tolerance factors, this huge number was narrowed to nearly 2700, and the corresponding decomposition energy and band gap calculations were performed for each one of them. However, the remaining nearly 26 300 perovskites, though not selected by the empirical criteria, could still hold valuable and potentially promising candidates. Therefore, an ML model has been trained on the DFT-calculated subset, which has been increased to 4181 to achieve molecular and elemental homogeneity in these data sets to predict and identify promising perovskites within the unexamined portion of the dataset. Remarkably, the ML approach identified 930 promising perovskites satisfying both the decomposition energy (≤0.025 eV per atom) and band gap (1.0 ≤ gap ≤ 2.0 eV) criteria. Among these, 20 perovskites were selected for further validation through DFT calculations, and a very nice agreement has been obtained between the predicted and calculated decomposition energy and band gap values. These findings highlight the effectiveness of ML in accelerating the discovery of materials with specific desirable properties.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 14","pages":" 7389-7398"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04218b?page=search","citationCount":"0","resultStr":"{\"title\":\"Discovering novel lead-free mixed cation hybrid halide perovskites via machine learning†\",\"authors\":\"Fatemeh Jamalinabijan, Somayyeh Alidoust, Gözde İniş Demir and Adem Tekin\",\"doi\":\"10.1039/D4CP04218B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In our recent study (S. Alidoust, F. Jamalinabijan and A. Tekin, <em>ACS Appl. Energy Mater.</em>, 2024, <strong>7</strong>, 785–798), a thorough computational screening using density functional theory (DFT) was conducted on mixed cation halide perovskites with a general formula of AA′BX<small><sub>3</sub></small>, aiming to identify promising lead-free candidates. Employment of 23 A/A′-cations, 29 B-ions, and 4 X-anions yielded approximately 29 000 possible perovskite combinations. However, while modern high-throughput DFT frameworks can handle large-scale calculations, treating the entire configurational space of 29 000 possible perovskite combinations remains computationally demanding. Leveraging machine learning (ML) approaches could provide a more efficient alternative for capturing this complexity. Therefore, by using two empirical criteria known as octahedral and tolerance factors, this huge number was narrowed to nearly 2700, and the corresponding decomposition energy and band gap calculations were performed for each one of them. However, the remaining nearly 26 300 perovskites, though not selected by the empirical criteria, could still hold valuable and potentially promising candidates. Therefore, an ML model has been trained on the DFT-calculated subset, which has been increased to 4181 to achieve molecular and elemental homogeneity in these data sets to predict and identify promising perovskites within the unexamined portion of the dataset. Remarkably, the ML approach identified 930 promising perovskites satisfying both the decomposition energy (≤0.025 eV per atom) and band gap (1.0 ≤ gap ≤ 2.0 eV) criteria. Among these, 20 perovskites were selected for further validation through DFT calculations, and a very nice agreement has been obtained between the predicted and calculated decomposition energy and band gap values. These findings highlight the effectiveness of ML in accelerating the discovery of materials with specific desirable properties.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 14\",\"pages\":\" 7389-7398\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-03-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04218b?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04218b\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04218b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
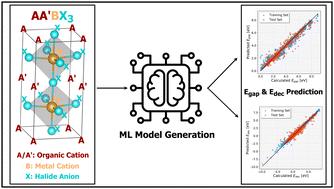
In our recent study (S. Alidoust, F. Jamalinabijan and A. Tekin, ACS Appl. Energy Mater., 2024, 7, 785–798), a thorough computational screening using density functional theory (DFT) was conducted on mixed cation halide perovskites with a general formula of AA′BX3, aiming to identify promising lead-free candidates. Employment of 23 A/A′-cations, 29 B-ions, and 4 X-anions yielded approximately 29 000 possible perovskite combinations. However, while modern high-throughput DFT frameworks can handle large-scale calculations, treating the entire configurational space of 29 000 possible perovskite combinations remains computationally demanding. Leveraging machine learning (ML) approaches could provide a more efficient alternative for capturing this complexity. Therefore, by using two empirical criteria known as octahedral and tolerance factors, this huge number was narrowed to nearly 2700, and the corresponding decomposition energy and band gap calculations were performed for each one of them. However, the remaining nearly 26 300 perovskites, though not selected by the empirical criteria, could still hold valuable and potentially promising candidates. Therefore, an ML model has been trained on the DFT-calculated subset, which has been increased to 4181 to achieve molecular and elemental homogeneity in these data sets to predict and identify promising perovskites within the unexamined portion of the dataset. Remarkably, the ML approach identified 930 promising perovskites satisfying both the decomposition energy (≤0.025 eV per atom) and band gap (1.0 ≤ gap ≤ 2.0 eV) criteria. Among these, 20 perovskites were selected for further validation through DFT calculations, and a very nice agreement has been obtained between the predicted and calculated decomposition energy and band gap values. These findings highlight the effectiveness of ML in accelerating the discovery of materials with specific desirable properties.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


