{"title":"高自旋分子中g矩阵分析的有效态相互作用方法","authors":"Antonio Cebreiro-Gallardo and David Casanova","doi":"10.1039/D4CP04511D","DOIUrl":null,"url":null,"abstract":"<p >We present an efficient state-interaction approach for evaluating <em>g</em>-shifts in high-spin molecular systems. Using a spin–orbit-coupled effective Hamiltonian with a restricted active space configuration interaction wavefunction, this method captures key excited-state contributions to <em>g</em>-shifts without requiring large orbital spaces, maintaining computational efficiency. Additionally, we introduce a property-driven algorithm to automatically select relevant orbitals, optimizing the active space selection. Application to diatomic and conjugated organic molecules demonstrates accuracy comparable to advanced methods, providing detailed insight into the origins of <em>g</em>-shifts. This methodology offers a flexible, efficient tool for exploring magnetic properties in complex molecules.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 14","pages":" 7093-7103"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04511d?page=search","citationCount":"0","resultStr":"{\"title\":\"Efficient state-interaction approach for the g-matrix analysis in high-spin molecules†\",\"authors\":\"Antonio Cebreiro-Gallardo and David Casanova\",\"doi\":\"10.1039/D4CP04511D\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We present an efficient state-interaction approach for evaluating <em>g</em>-shifts in high-spin molecular systems. Using a spin–orbit-coupled effective Hamiltonian with a restricted active space configuration interaction wavefunction, this method captures key excited-state contributions to <em>g</em>-shifts without requiring large orbital spaces, maintaining computational efficiency. Additionally, we introduce a property-driven algorithm to automatically select relevant orbitals, optimizing the active space selection. Application to diatomic and conjugated organic molecules demonstrates accuracy comparable to advanced methods, providing detailed insight into the origins of <em>g</em>-shifts. This methodology offers a flexible, efficient tool for exploring magnetic properties in complex molecules.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 14\",\"pages\":\" 7093-7103\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-03-14\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d4cp04511d?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04511d\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04511d","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Efficient state-interaction approach for the g-matrix analysis in high-spin molecules†
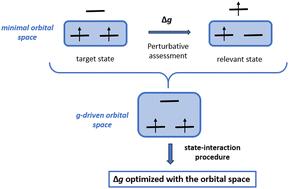
We present an efficient state-interaction approach for evaluating g-shifts in high-spin molecular systems. Using a spin–orbit-coupled effective Hamiltonian with a restricted active space configuration interaction wavefunction, this method captures key excited-state contributions to g-shifts without requiring large orbital spaces, maintaining computational efficiency. Additionally, we introduce a property-driven algorithm to automatically select relevant orbitals, optimizing the active space selection. Application to diatomic and conjugated organic molecules demonstrates accuracy comparable to advanced methods, providing detailed insight into the origins of g-shifts. This methodology offers a flexible, efficient tool for exploring magnetic properties in complex molecules.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


