Sanni Översti, Ariane Weber, Viktor Baran, Bärbel Kieninger, Alexander Dilthey, Torsten Houwaart, Andreas Walker, Wulf Schneider-Brachert, Denise Kühnert
{"title":"以三个贝叶斯系统动力学案例为例分析新冠肺炎在德国的进化和流行动力学。","authors":"Sanni Översti, Ariane Weber, Viktor Baran, Bärbel Kieninger, Alexander Dilthey, Torsten Houwaart, Andreas Walker, Wulf Schneider-Brachert, Denise Kühnert","doi":"10.1177/11779322251321065","DOIUrl":null,"url":null,"abstract":"<p><p>The importance of genomic surveillance strategies for pathogens has been particularly evident during the coronavirus disease 2019 (COVID-19) pandemic, as genomic data from the causative agent, severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), have guided public health decisions worldwide. Bayesian phylodynamic inference, integrating epidemiology and evolutionary biology, has become an essential tool in genomic epidemiological surveillance. It enables the estimation of epidemiological parameters, such as the reproductive number, from pathogen sequence data alone. Despite the phylodynamic approach being widely adopted, the abundance of phylodynamic models often makes it challenging to select the appropriate model for specific research questions. This article illustrates the application of phylodynamic birth-death-sampling models in public health using genomic data, with a focus on SARS-CoV-2. Targeting researchers less familiar with phylodynamics, it introduces a comprehensive workflow, including the conceptualisation of a research study and detailed steps for data preprocessing and postprocessing. In addition, we demonstrate the versatility of birth-death-sampling models through three case studies from Germany, utilising the BEAST2 software and its model implementations. Each case study addresses a distinct research question relevant not only to SARS-CoV-2 but also to other pathogens: Case study 1 finds traces of a superspreading event at the start of an early outbreak, exemplifying how simple models for genomic data can provide information that would otherwise only be accessible through extensive contact tracing. Case study 2 compares transmission dynamics in a nosocomial outbreak to community transmission, highlighting distinct dynamics through integrative analysis. Case study 3 investigates whether local transmission patterns align with national trends, demonstrating how phylodynamic models can disentangle complex population substructure with little additional information. For each case study, we emphasise critical points where model assumptions and data properties may misalign and outline appropriate validation assessments. Overall, we aim to provide researchers with examples on using birth-death-sampling models in genomic epidemiology, balancing theoretical and practical aspects.</p>","PeriodicalId":9065,"journal":{"name":"Bioinformatics and Biology Insights","volume":"19 ","pages":"11779322251321065"},"PeriodicalIF":2.4000,"publicationDate":"2025-03-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11898094/pdf/","citationCount":"0","resultStr":"{\"title\":\"Evolutionary and epidemic dynamics of COVID-19 in Germany exemplified by three Bayesian phylodynamic case studies.\",\"authors\":\"Sanni Översti, Ariane Weber, Viktor Baran, Bärbel Kieninger, Alexander Dilthey, Torsten Houwaart, Andreas Walker, Wulf Schneider-Brachert, Denise Kühnert\",\"doi\":\"10.1177/11779322251321065\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The importance of genomic surveillance strategies for pathogens has been particularly evident during the coronavirus disease 2019 (COVID-19) pandemic, as genomic data from the causative agent, severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), have guided public health decisions worldwide. Bayesian phylodynamic inference, integrating epidemiology and evolutionary biology, has become an essential tool in genomic epidemiological surveillance. It enables the estimation of epidemiological parameters, such as the reproductive number, from pathogen sequence data alone. Despite the phylodynamic approach being widely adopted, the abundance of phylodynamic models often makes it challenging to select the appropriate model for specific research questions. This article illustrates the application of phylodynamic birth-death-sampling models in public health using genomic data, with a focus on SARS-CoV-2. Targeting researchers less familiar with phylodynamics, it introduces a comprehensive workflow, including the conceptualisation of a research study and detailed steps for data preprocessing and postprocessing. In addition, we demonstrate the versatility of birth-death-sampling models through three case studies from Germany, utilising the BEAST2 software and its model implementations. Each case study addresses a distinct research question relevant not only to SARS-CoV-2 but also to other pathogens: Case study 1 finds traces of a superspreading event at the start of an early outbreak, exemplifying how simple models for genomic data can provide information that would otherwise only be accessible through extensive contact tracing. Case study 2 compares transmission dynamics in a nosocomial outbreak to community transmission, highlighting distinct dynamics through integrative analysis. Case study 3 investigates whether local transmission patterns align with national trends, demonstrating how phylodynamic models can disentangle complex population substructure with little additional information. For each case study, we emphasise critical points where model assumptions and data properties may misalign and outline appropriate validation assessments. Overall, we aim to provide researchers with examples on using birth-death-sampling models in genomic epidemiology, balancing theoretical and practical aspects.</p>\",\"PeriodicalId\":9065,\"journal\":{\"name\":\"Bioinformatics and Biology Insights\",\"volume\":\"19 \",\"pages\":\"11779322251321065\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2025-03-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11898094/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Bioinformatics and Biology Insights\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1177/11779322251321065\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Bioinformatics and Biology Insights","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1177/11779322251321065","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Evolutionary and epidemic dynamics of COVID-19 in Germany exemplified by three Bayesian phylodynamic case studies.
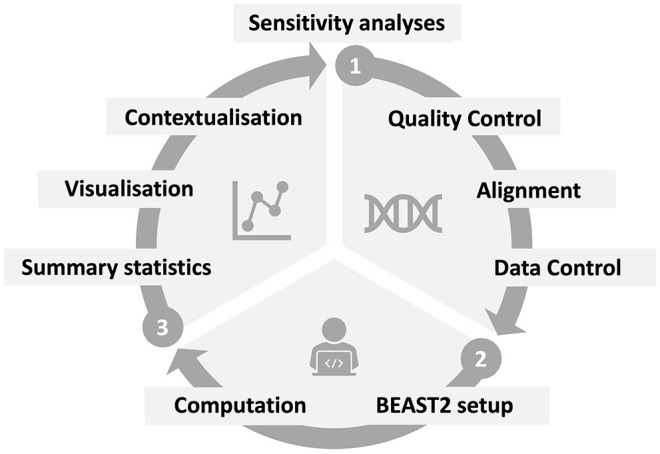
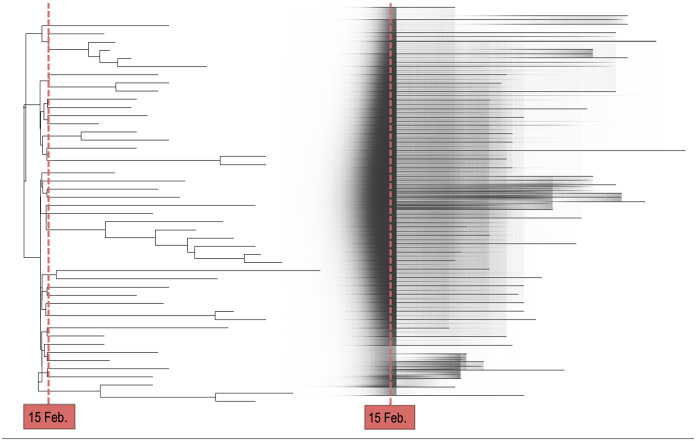
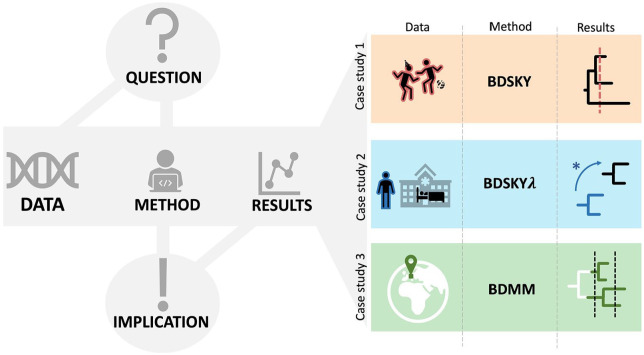
The importance of genomic surveillance strategies for pathogens has been particularly evident during the coronavirus disease 2019 (COVID-19) pandemic, as genomic data from the causative agent, severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), have guided public health decisions worldwide. Bayesian phylodynamic inference, integrating epidemiology and evolutionary biology, has become an essential tool in genomic epidemiological surveillance. It enables the estimation of epidemiological parameters, such as the reproductive number, from pathogen sequence data alone. Despite the phylodynamic approach being widely adopted, the abundance of phylodynamic models often makes it challenging to select the appropriate model for specific research questions. This article illustrates the application of phylodynamic birth-death-sampling models in public health using genomic data, with a focus on SARS-CoV-2. Targeting researchers less familiar with phylodynamics, it introduces a comprehensive workflow, including the conceptualisation of a research study and detailed steps for data preprocessing and postprocessing. In addition, we demonstrate the versatility of birth-death-sampling models through three case studies from Germany, utilising the BEAST2 software and its model implementations. Each case study addresses a distinct research question relevant not only to SARS-CoV-2 but also to other pathogens: Case study 1 finds traces of a superspreading event at the start of an early outbreak, exemplifying how simple models for genomic data can provide information that would otherwise only be accessible through extensive contact tracing. Case study 2 compares transmission dynamics in a nosocomial outbreak to community transmission, highlighting distinct dynamics through integrative analysis. Case study 3 investigates whether local transmission patterns align with national trends, demonstrating how phylodynamic models can disentangle complex population substructure with little additional information. For each case study, we emphasise critical points where model assumptions and data properties may misalign and outline appropriate validation assessments. Overall, we aim to provide researchers with examples on using birth-death-sampling models in genomic epidemiology, balancing theoretical and practical aspects.
期刊介绍:
Bioinformatics and Biology Insights is an open access, peer-reviewed journal that considers articles on bioinformatics methods and their applications which must pertain to biological insights. All papers should be easily amenable to biologists and as such help bridge the gap between theories and applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


