Shubham Agarwal, Hussam Alkaissi, Karel Pacak, Jorge Esteban Mosquera Izurieta, Alan P Dackiw, Sarah C Oltmann, Fiemu Nwariaku, Liwei Jia, Mary Grace Roden, Oksana Hamidi
{"title":"复合嗜铬细胞瘤-副神经节瘤伴神经节神经瘤:双中心临床经验。","authors":"Shubham Agarwal, Hussam Alkaissi, Karel Pacak, Jorge Esteban Mosquera Izurieta, Alan P Dackiw, Sarah C Oltmann, Fiemu Nwariaku, Liwei Jia, Mary Grace Roden, Oksana Hamidi","doi":"10.1210/jendso/bvaf036","DOIUrl":null,"url":null,"abstract":"<p><strong>Context: </strong>Cells derived from neural crest populate several organs. A particular precursor cell, sympathogonia, gives rise to pheochromoblasts and neuroblasts. Due to common origin, tumors originating from pheochromoblasts, such as pheochromocytoma (PHEO) and paraganglioma (PGL), may rarely coexist with ganglioneuroma (GN).</p><p><strong>Objective: </strong>We evaluated clinical, biochemical, and radiological characteristics of patients with composite PHEO/PGL and GN (PPGL-GN) and compared them to patients with PHEO.</p><p><strong>Methods: </strong>In this retrospective, dual-center, observational, case-control study, we identified patients with PPGL-GN. Similarly, we identified a control group of patients with PHEO who underwent laparoscopic adrenalectomy. All diagnoses were confirmed on histology. Descriptive statistics were used to summarize demographic and clinical data.</p><p><strong>Results: </strong>We identified 19 consecutive patients with PPGL-GN and 86 patients with PHEO. Patients with PPGL-GN, compared to those with PHEO, were younger (aged 46.0 vs 50.8 years; <i>P</i> = .03), had higher rate of underlying genetic disorders (47.4% vs 23.2%; <i>P</i> = .03), and had fewer functioning tumors (89.5% vs 100%; <i>P</i> = .002). There was no difference in the median radiological tumor size or the precontrast computed tomography density. Disease recurrence (at another site) was noted in 15.8% of PPGL-GN patients who had a median follow up of 14.6 months, as opposed to no disease recurrence in patients with PHEO. There was no documented recurrence at the tumor bed and no metastasis in both groups.</p><p><strong>Conclusion: </strong>Patients with PPGL-GN were younger and had a higher occurrence of underlying genetic disorders compared to PHEO. However, PPGL-GN was radiologically indistinguishable from PHEO. The higher observed disease recurrence of PPGL-GN reinforces vigilant postoperative follow-up.</p>","PeriodicalId":17334,"journal":{"name":"Journal of the Endocrine Society","volume":"9 4","pages":"bvaf036"},"PeriodicalIF":3.1000,"publicationDate":"2025-02-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11893535/pdf/","citationCount":"0","resultStr":"{\"title\":\"Composite Pheochromocytoma-Paraganglioma With Ganglioneuroma: A Dual-Center Clinical Experience.\",\"authors\":\"Shubham Agarwal, Hussam Alkaissi, Karel Pacak, Jorge Esteban Mosquera Izurieta, Alan P Dackiw, Sarah C Oltmann, Fiemu Nwariaku, Liwei Jia, Mary Grace Roden, Oksana Hamidi\",\"doi\":\"10.1210/jendso/bvaf036\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Context: </strong>Cells derived from neural crest populate several organs. A particular precursor cell, sympathogonia, gives rise to pheochromoblasts and neuroblasts. Due to common origin, tumors originating from pheochromoblasts, such as pheochromocytoma (PHEO) and paraganglioma (PGL), may rarely coexist with ganglioneuroma (GN).</p><p><strong>Objective: </strong>We evaluated clinical, biochemical, and radiological characteristics of patients with composite PHEO/PGL and GN (PPGL-GN) and compared them to patients with PHEO.</p><p><strong>Methods: </strong>In this retrospective, dual-center, observational, case-control study, we identified patients with PPGL-GN. Similarly, we identified a control group of patients with PHEO who underwent laparoscopic adrenalectomy. All diagnoses were confirmed on histology. Descriptive statistics were used to summarize demographic and clinical data.</p><p><strong>Results: </strong>We identified 19 consecutive patients with PPGL-GN and 86 patients with PHEO. Patients with PPGL-GN, compared to those with PHEO, were younger (aged 46.0 vs 50.8 years; <i>P</i> = .03), had higher rate of underlying genetic disorders (47.4% vs 23.2%; <i>P</i> = .03), and had fewer functioning tumors (89.5% vs 100%; <i>P</i> = .002). There was no difference in the median radiological tumor size or the precontrast computed tomography density. Disease recurrence (at another site) was noted in 15.8% of PPGL-GN patients who had a median follow up of 14.6 months, as opposed to no disease recurrence in patients with PHEO. There was no documented recurrence at the tumor bed and no metastasis in both groups.</p><p><strong>Conclusion: </strong>Patients with PPGL-GN were younger and had a higher occurrence of underlying genetic disorders compared to PHEO. However, PPGL-GN was radiologically indistinguishable from PHEO. The higher observed disease recurrence of PPGL-GN reinforces vigilant postoperative follow-up.</p>\",\"PeriodicalId\":17334,\"journal\":{\"name\":\"Journal of the Endocrine Society\",\"volume\":\"9 4\",\"pages\":\"bvaf036\"},\"PeriodicalIF\":3.1000,\"publicationDate\":\"2025-02-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11893535/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the Endocrine Society\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1210/jendso/bvaf036\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/3/3 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q2\",\"JCRName\":\"ENDOCRINOLOGY & METABOLISM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Endocrine Society","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1210/jendso/bvaf036","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/3 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
摘要
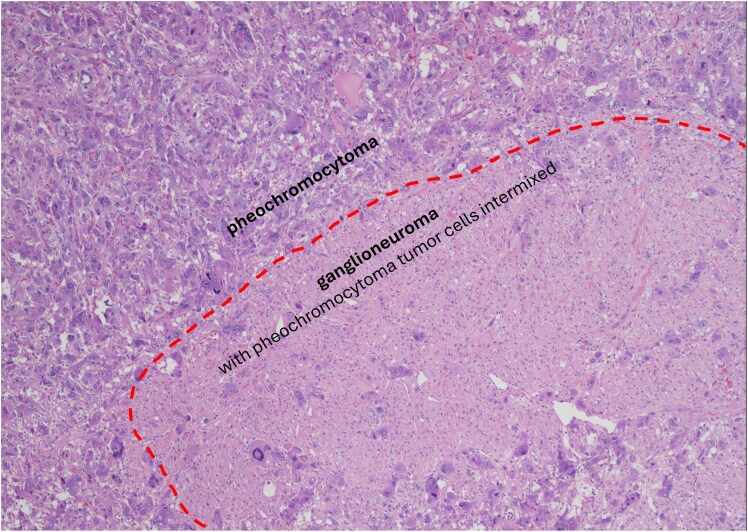
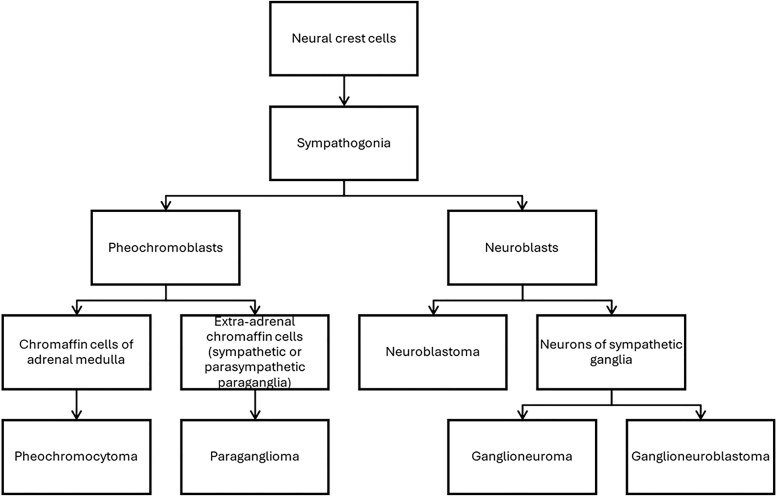
背景神经嵴衍生的细胞可填充多个器官。一种特殊的前体细胞--嗜铬细胞(sympathogonia)会产生嗜铬细胞和神经母细胞。由于起源相同,嗜铬细胞瘤(PHEO)和副神经节瘤(PGL)等起源于嗜铬细胞的肿瘤很少与神经节细胞瘤(GN)同时存在:我们评估了PHEO/PGL和GN复合型(PPGL-GN)患者的临床、生化和放射学特征,并将其与PHEO患者进行了比较:在这项回顾性、双中心、观察性、病例对照研究中,我们确定了 PPGL-GN 患者。同样,我们还确定了一组接受腹腔镜肾上腺切除术的 PHEO 患者作为对照组。所有诊断均经组织学确诊。我们使用描述性统计来总结人口统计学和临床数据:结果:我们连续发现了19例PPGL-GN患者和86例PHEO患者。与PHEO患者相比,PPGL-GN患者更年轻(46.0岁 vs 50.8岁;P = .03),潜在遗传疾病的比例更高(47.4% vs 23.2%;P = .03),功能性肿瘤更少(89.5% vs 100%;P = .002)。中位放射学肿瘤大小或对比前计算机断层扫描密度没有差异。15.8%的PPGL-GN患者在中位随访14.6个月后发现疾病复发(在其他部位),而PHEO患者则没有疾病复发。两组患者均无肿瘤床复发和转移的记录:结论:与PHEO相比,PPGL-GN患者更年轻,潜在遗传疾病的发生率更高。然而,PPGL-GN 在放射学上与 PHEO 没有区别。观察到PPGL-GN的疾病复发率较高,因此需要提高警惕进行术后随访。
Composite Pheochromocytoma-Paraganglioma With Ganglioneuroma: A Dual-Center Clinical Experience.
Context: Cells derived from neural crest populate several organs. A particular precursor cell, sympathogonia, gives rise to pheochromoblasts and neuroblasts. Due to common origin, tumors originating from pheochromoblasts, such as pheochromocytoma (PHEO) and paraganglioma (PGL), may rarely coexist with ganglioneuroma (GN).
Objective: We evaluated clinical, biochemical, and radiological characteristics of patients with composite PHEO/PGL and GN (PPGL-GN) and compared them to patients with PHEO.
Methods: In this retrospective, dual-center, observational, case-control study, we identified patients with PPGL-GN. Similarly, we identified a control group of patients with PHEO who underwent laparoscopic adrenalectomy. All diagnoses were confirmed on histology. Descriptive statistics were used to summarize demographic and clinical data.
Results: We identified 19 consecutive patients with PPGL-GN and 86 patients with PHEO. Patients with PPGL-GN, compared to those with PHEO, were younger (aged 46.0 vs 50.8 years; P = .03), had higher rate of underlying genetic disorders (47.4% vs 23.2%; P = .03), and had fewer functioning tumors (89.5% vs 100%; P = .002). There was no difference in the median radiological tumor size or the precontrast computed tomography density. Disease recurrence (at another site) was noted in 15.8% of PPGL-GN patients who had a median follow up of 14.6 months, as opposed to no disease recurrence in patients with PHEO. There was no documented recurrence at the tumor bed and no metastasis in both groups.
Conclusion: Patients with PPGL-GN were younger and had a higher occurrence of underlying genetic disorders compared to PHEO. However, PPGL-GN was radiologically indistinguishable from PHEO. The higher observed disease recurrence of PPGL-GN reinforces vigilant postoperative follow-up.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


