{"title":"氧化锌中外源性铁缺陷的光主动振动","authors":"Alexey N. Kislov and Anatoly F. Zatsepin","doi":"10.1039/D5CP00477B","DOIUrl":null,"url":null,"abstract":"<p >In the paper, using Fe<small><sup>3+</sup></small> ions in a non-centrosymmetric ZnO lattice as an example, we present a theoretical study of impurity-induced vibrations. The modeling was carried out within the framework of density functional theory using the generalized gradient approximation and the potential-based method. The <em>C</em><small><sub>3v</sub></small> lattice distortions around a trivalent impurity were computed. Independent calculation methods give similar results, which indicates their reliability. We calculated local symmetrized phonon densities of states in Fe-doped ZnO and determined the frequencies of the impurity vibrations of different symmetry types induced by charged Fe ions. The results of lattice-dynamic calculations were used to interpret the structure of the phonon sideband that accompanies the zero-phonon line in the polarized emission spectra associated with intracenter transitions of Fe<small><sup>3+</sup></small>. We believe that the approach used allows us to objectively evaluate the contribution of charged impurities with a weak electron–phonon coupling and surrounding ions to the formation of the main peaks observed in the vibronic spectrum of crystals.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 13","pages":" 6724-6729"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Optically active vibrations of extrinsic iron defects in zinc oxide\",\"authors\":\"Alexey N. Kislov and Anatoly F. Zatsepin\",\"doi\":\"10.1039/D5CP00477B\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >In the paper, using Fe<small><sup>3+</sup></small> ions in a non-centrosymmetric ZnO lattice as an example, we present a theoretical study of impurity-induced vibrations. The modeling was carried out within the framework of density functional theory using the generalized gradient approximation and the potential-based method. The <em>C</em><small><sub>3v</sub></small> lattice distortions around a trivalent impurity were computed. Independent calculation methods give similar results, which indicates their reliability. We calculated local symmetrized phonon densities of states in Fe-doped ZnO and determined the frequencies of the impurity vibrations of different symmetry types induced by charged Fe ions. The results of lattice-dynamic calculations were used to interpret the structure of the phonon sideband that accompanies the zero-phonon line in the polarized emission spectra associated with intracenter transitions of Fe<small><sup>3+</sup></small>. We believe that the approach used allows us to objectively evaluate the contribution of charged impurities with a weak electron–phonon coupling and surrounding ions to the formation of the main peaks observed in the vibronic spectrum of crystals.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 13\",\"pages\":\" 6724-6729\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-03-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00477b\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00477b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Optically active vibrations of extrinsic iron defects in zinc oxide
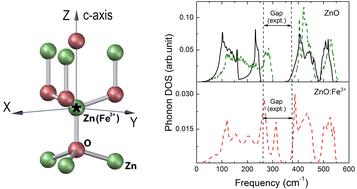
In the paper, using Fe3+ ions in a non-centrosymmetric ZnO lattice as an example, we present a theoretical study of impurity-induced vibrations. The modeling was carried out within the framework of density functional theory using the generalized gradient approximation and the potential-based method. The C3v lattice distortions around a trivalent impurity were computed. Independent calculation methods give similar results, which indicates their reliability. We calculated local symmetrized phonon densities of states in Fe-doped ZnO and determined the frequencies of the impurity vibrations of different symmetry types induced by charged Fe ions. The results of lattice-dynamic calculations were used to interpret the structure of the phonon sideband that accompanies the zero-phonon line in the polarized emission spectra associated with intracenter transitions of Fe3+. We believe that the approach used allows us to objectively evaluate the contribution of charged impurities with a weak electron–phonon coupling and surrounding ions to the formation of the main peaks observed in the vibronic spectrum of crystals.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


