{"title":"组蛋白去乙酰化酶8的去乙酰化机制:来自QM/MM MP2计算的见解","authors":"Rui Lai and Hui Li","doi":"10.1039/D5CP00002E","DOIUrl":null,"url":null,"abstract":"<p >Understanding the catalytic mechanism of histone deacetylases can greatly benefit the development of targeted therapies that are safe and effective. Combined quantum mechanical and molecular mechanical (QM/MM) Møller–Plesset second-order perturbation theory (MP2) geometry optimizations are performed to investigate the catalytic mechanism of the deacetylation reaction of a tetrapeptide catalyzed by human Histone Deacetylase 8. A three-step catalytic mechanism is identified: the first step is the formation of a negatively charged tetrahedral intermediate <em>via</em> nucleophilic addition of the activated water to the amide C atom and a proton transfer from the water to His143; the second step is the formation of a neutral tetrahedral intermediate with an elongated amide C–N bond <em>via</em> a proton transfer from His143 to the amide N atom. The third step is the complete cleavage of the amide C–N bond, accompanied by a proton transfer from the newly formed carboxylic group of the neutral tetrahedral intermediate to His142. These three steps have similar computed energy barriers, with the second step having the highest calculated activation free energy of 19.6 kcal mol<small><sup>−1</sup></small>. When there is no potassium ion at site 1, the calculated activation free energy is 17.7 kcal mol<small><sup>−1</sup></small>. Both values are in good agreement with an experimental value of 17.5 kcal mol<small><sup>−1</sup></small>. Their difference implies that there would be a 25-fold increase in the enzyme's activity, in line with experiments. The solvent hydrogen–deuterium kinetic isotope effect was computed to be ∼3.8 for the second step in both cases. It is also found that the energy barriers are significantly and systematically higher on the QM/MM B3LYP and QM/MM B3LYP-D3 potential energy surfaces. In particular, the QM/MM B3LYP and B3LYP-D3 methods fail to predict the neutral tetrahedral intermediate and a meaningful transition state for the third step, leading to a two-step mechanism. With a sufficiently large basis set such as aug-cc-pVDZ, QM/MM M05-2X, M06-2X, M06, and MN15 methods can give results much closer to the QM/MM MP2 method. However, when a smaller basis set such as 6-31G* is used, these methods can lead to errors as large as 10 kcal mol<small><sup>−1</sup></small> on the reaction pathway. These results highlight the importance of using accurate QM methods in the computational study of enzyme catalysis.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 14","pages":" 7120-7138"},"PeriodicalIF":2.9000,"publicationDate":"2025-03-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00002e?page=search","citationCount":"0","resultStr":"{\"title\":\"Deacetylation mechanism of histone deacetylase 8: insights from QM/MM MP2 calculations†\",\"authors\":\"Rui Lai and Hui Li\",\"doi\":\"10.1039/D5CP00002E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Understanding the catalytic mechanism of histone deacetylases can greatly benefit the development of targeted therapies that are safe and effective. Combined quantum mechanical and molecular mechanical (QM/MM) Møller–Plesset second-order perturbation theory (MP2) geometry optimizations are performed to investigate the catalytic mechanism of the deacetylation reaction of a tetrapeptide catalyzed by human Histone Deacetylase 8. A three-step catalytic mechanism is identified: the first step is the formation of a negatively charged tetrahedral intermediate <em>via</em> nucleophilic addition of the activated water to the amide C atom and a proton transfer from the water to His143; the second step is the formation of a neutral tetrahedral intermediate with an elongated amide C–N bond <em>via</em> a proton transfer from His143 to the amide N atom. The third step is the complete cleavage of the amide C–N bond, accompanied by a proton transfer from the newly formed carboxylic group of the neutral tetrahedral intermediate to His142. These three steps have similar computed energy barriers, with the second step having the highest calculated activation free energy of 19.6 kcal mol<small><sup>−1</sup></small>. When there is no potassium ion at site 1, the calculated activation free energy is 17.7 kcal mol<small><sup>−1</sup></small>. Both values are in good agreement with an experimental value of 17.5 kcal mol<small><sup>−1</sup></small>. Their difference implies that there would be a 25-fold increase in the enzyme's activity, in line with experiments. The solvent hydrogen–deuterium kinetic isotope effect was computed to be ∼3.8 for the second step in both cases. It is also found that the energy barriers are significantly and systematically higher on the QM/MM B3LYP and QM/MM B3LYP-D3 potential energy surfaces. In particular, the QM/MM B3LYP and B3LYP-D3 methods fail to predict the neutral tetrahedral intermediate and a meaningful transition state for the third step, leading to a two-step mechanism. With a sufficiently large basis set such as aug-cc-pVDZ, QM/MM M05-2X, M06-2X, M06, and MN15 methods can give results much closer to the QM/MM MP2 method. However, when a smaller basis set such as 6-31G* is used, these methods can lead to errors as large as 10 kcal mol<small><sup>−1</sup></small> on the reaction pathway. These results highlight the importance of using accurate QM methods in the computational study of enzyme catalysis.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 14\",\"pages\":\" 7120-7138\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-03-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00002e?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00002e\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00002e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Deacetylation mechanism of histone deacetylase 8: insights from QM/MM MP2 calculations†
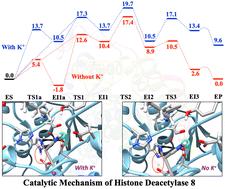
Understanding the catalytic mechanism of histone deacetylases can greatly benefit the development of targeted therapies that are safe and effective. Combined quantum mechanical and molecular mechanical (QM/MM) Møller–Plesset second-order perturbation theory (MP2) geometry optimizations are performed to investigate the catalytic mechanism of the deacetylation reaction of a tetrapeptide catalyzed by human Histone Deacetylase 8. A three-step catalytic mechanism is identified: the first step is the formation of a negatively charged tetrahedral intermediate via nucleophilic addition of the activated water to the amide C atom and a proton transfer from the water to His143; the second step is the formation of a neutral tetrahedral intermediate with an elongated amide C–N bond via a proton transfer from His143 to the amide N atom. The third step is the complete cleavage of the amide C–N bond, accompanied by a proton transfer from the newly formed carboxylic group of the neutral tetrahedral intermediate to His142. These three steps have similar computed energy barriers, with the second step having the highest calculated activation free energy of 19.6 kcal mol−1. When there is no potassium ion at site 1, the calculated activation free energy is 17.7 kcal mol−1. Both values are in good agreement with an experimental value of 17.5 kcal mol−1. Their difference implies that there would be a 25-fold increase in the enzyme's activity, in line with experiments. The solvent hydrogen–deuterium kinetic isotope effect was computed to be ∼3.8 for the second step in both cases. It is also found that the energy barriers are significantly and systematically higher on the QM/MM B3LYP and QM/MM B3LYP-D3 potential energy surfaces. In particular, the QM/MM B3LYP and B3LYP-D3 methods fail to predict the neutral tetrahedral intermediate and a meaningful transition state for the third step, leading to a two-step mechanism. With a sufficiently large basis set such as aug-cc-pVDZ, QM/MM M05-2X, M06-2X, M06, and MN15 methods can give results much closer to the QM/MM MP2 method. However, when a smaller basis set such as 6-31G* is used, these methods can lead to errors as large as 10 kcal mol−1 on the reaction pathway. These results highlight the importance of using accurate QM methods in the computational study of enzyme catalysis.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


