利用DFT和蒙特卡罗方法研究真空条件下AuPd合金纳米颗粒结构
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
摘要
AuPd是一种常用于催化的混相金属合金。负载型AuPd催化剂,在高Au/Pd比下,形成单原子合金(SAAs),已被证明可以提高许多催化反应的速率和/或选择性,包括(脱)氢化,氢解,C-C和C-O偶联反应。虽然许多计算研究已经检查了AuPd结构的稳定性(混相合金中的原子排列),但大多数研究都集中在通用合金上,而不是SAAs,而那些密切研究SAAs的研究则集中在单晶表面上。在这项工作中,我们使用密度泛函理论(DFT)来计算201原子截断八面体纳米粒子模型中的交换能(将Au原子与Pd原子交换),重点关注具有高Au/Pd比的粒子。我们将这些交换能计算为Pd在纳米颗粒中的位置、这些交换点附近的Pd原子数量以及纳米颗粒中Pd的总含量的函数。这些dft计算的交换能也被用于告知简单的基于物理的模型(与聚类扩展或神经网络模型相反),这些模型与dft计算的值具有相对较少的回归参数,表现出良好的一致性。然后将这些模型应用到蒙特卡罗(MC)模拟中,以预测纳米颗粒结构作为组成和温度的函数。结果表明,钯倾向于在纳米颗粒的亚表面,钯倾向于在Au中与自身隔离。这两种观察结果都与先前对单晶系统的实验和计算研究相吻合。我们还表明,纳米粒子的整体组成通过改变系统的电子性质(例如费米能级)来影响交换能,这与Pd比Au少一个价电子有关。MC模拟表明,在真空中,Pd在约20 mol % Pd (298 K)时开始填充这些~ 2 nm纳米颗粒的表面,并且SAA应用所需的Pd表面单体数量在接近40 mol % Pd时达到最大值。随着温度的升高,Pd在地表更为普遍,但温度的影响相对较小。虽然已知AuPd结构在反应气体(如CO或O2)存在下会发生变化,但这些研究表征了基本的热力学排列,可用于了解催化剂表征和反应研究过程中的表面重组。本文章由计算机程序翻译,如有差异,请以英文原文为准。
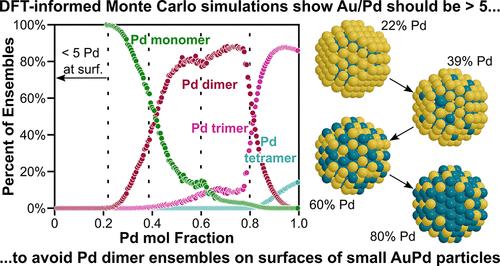
Understanding AuPd Alloy Nanoparticle Structure under Vacuum Using DFT and Monte Carlo Methods
AuPd is a miscible metal alloy that is often used in catalysis. Supported AuPd catalysts, at high Au/Pd ratios, form single-atom alloys (SAAs) that have been shown to enhance rates and/or selectivities for many catalytic reactions, including (de)hydrogenations, hydrogenolysis, and C–C and C–O coupling reactions. While many computational studies have examined the stability of AuPd structures (the arrangement of atoms within the miscible alloy), most focused on generic alloys rather than SAAs and those that have closely investigated SAAs focused on single crystal surfaces. In this work, we use density functional theory (DFT) to calculate exchange energies (swapping an Au atom with a Pd atom) in a 201-atom truncated octahedral nanoparticle model with a focus on particles with high Au/Pd ratios. We calculate these exchange energies as a function of Pd location within the nanoparticle, the number of Pd atoms neighboring and near those exchange sites, and the total Pd content in the nanoparticle. These DFT-calculated exchange energies are also used to inform simple physics-based models (in contrast to cluster expansion or neural network models) that show good agreement with DFT-calculated values with relatively few regressed parameters. These models are then implemented into Monte Carlo (MC) simulations to predict the nanoparticle structure as a function of composition and temperature. The results show that Pd prefers to be in the subsurface of nanoparticles and that Pd prefers to be isolated from itself within Au. Both observations agree well with prior experimental and computational studies of single-crystal systems. We also show that the overall composition of the nanoparticle influences exchange energies by changing the electronic properties (e.g., Fermi level) of the system, which is relevant as Pd has one fewer valence electron than Au. MC simulations show that, in a vacuum, Pd begins to populate the surface of these ∼2 nm nanoparticles at around 20 mol % Pd (at 298 K) and that the number of Pd surface monomers, desired for SAA applications, goes through a maximum near 40 mol % Pd. As the temperature increases, Pd is more prevalent at the surface, but the influence of temperature is relatively muted. While AuPd structures are known to change in the presence of reactive gases (e.g., CO or O2), these studies characterize the baseline thermodynamic arrangements that can be used to understand surface restructuring during catalyst characterization and reaction studies.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


