{"title":"利用卷积神经网络从转录物丰度推断蛋白质。","authors":"Patrick Maximilian Schwehn, Pascal Falter-Braun","doi":"10.1186/s13040-025-00434-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Although transcript abundance is often used as a proxy for protein abundance, it is an unreliable predictor. As proteins execute biological functions and their expression levels influence phenotypic outcomes, we developed a convolutional neural network (CNN) to predict protein abundances from mRNA abundances, protein sequence, and mRNA sequence in Homo sapiens (H. sapiens) and the reference plant Arabidopsis thaliana (A. thaliana).</p><p><strong>Results: </strong>After hyperparameter optimization and initial data exploration, we implemented distinct training modules for value-based and sequence-based data. By analyzing the learned weights, we revealed common and organism-specific sequence features that influence protein-to-mRNA ratios (PTRs), including known and putative sequence motifs. Adding condition-specific protein interaction information identified genes correlated with many PTRs but did not improve predictions, likely due to insufficient data. The integrated model predicted protein abundance on unseen genes with a coefficient of determination (r<sup>2</sup>) of 0.30 in H. sapiens and 0.32 in A. thaliana.</p><p><strong>Conclusions: </strong>For H. sapiens, our model improves prediction performance by nearly 50% compared to previous sequence-based approaches, and for A. thaliana it represents the first model of its kind. The model's learned motifs recapitulate known regulatory elements, supporting its utility in systems-level and hypothesis-driven research approaches related to protein regulation.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"18 1","pages":"18"},"PeriodicalIF":6.1000,"publicationDate":"2025-02-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11866710/pdf/","citationCount":"0","resultStr":"{\"title\":\"Inferring protein from transcript abundances using convolutional neural networks.\",\"authors\":\"Patrick Maximilian Schwehn, Pascal Falter-Braun\",\"doi\":\"10.1186/s13040-025-00434-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Although transcript abundance is often used as a proxy for protein abundance, it is an unreliable predictor. As proteins execute biological functions and their expression levels influence phenotypic outcomes, we developed a convolutional neural network (CNN) to predict protein abundances from mRNA abundances, protein sequence, and mRNA sequence in Homo sapiens (H. sapiens) and the reference plant Arabidopsis thaliana (A. thaliana).</p><p><strong>Results: </strong>After hyperparameter optimization and initial data exploration, we implemented distinct training modules for value-based and sequence-based data. By analyzing the learned weights, we revealed common and organism-specific sequence features that influence protein-to-mRNA ratios (PTRs), including known and putative sequence motifs. Adding condition-specific protein interaction information identified genes correlated with many PTRs but did not improve predictions, likely due to insufficient data. The integrated model predicted protein abundance on unseen genes with a coefficient of determination (r<sup>2</sup>) of 0.30 in H. sapiens and 0.32 in A. thaliana.</p><p><strong>Conclusions: </strong>For H. sapiens, our model improves prediction performance by nearly 50% compared to previous sequence-based approaches, and for A. thaliana it represents the first model of its kind. The model's learned motifs recapitulate known regulatory elements, supporting its utility in systems-level and hypothesis-driven research approaches related to protein regulation.</p>\",\"PeriodicalId\":48947,\"journal\":{\"name\":\"Biodata Mining\",\"volume\":\"18 1\",\"pages\":\"18\"},\"PeriodicalIF\":6.1000,\"publicationDate\":\"2025-02-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11866710/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biodata Mining\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13040-025-00434-z\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-025-00434-z","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Inferring protein from transcript abundances using convolutional neural networks.
Background: Although transcript abundance is often used as a proxy for protein abundance, it is an unreliable predictor. As proteins execute biological functions and their expression levels influence phenotypic outcomes, we developed a convolutional neural network (CNN) to predict protein abundances from mRNA abundances, protein sequence, and mRNA sequence in Homo sapiens (H. sapiens) and the reference plant Arabidopsis thaliana (A. thaliana).
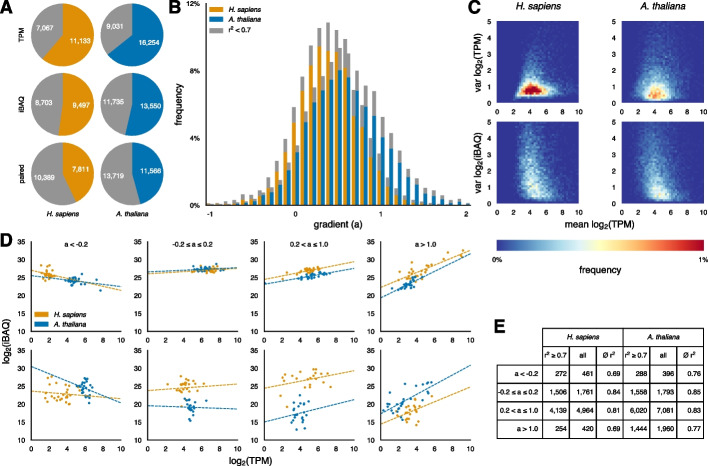
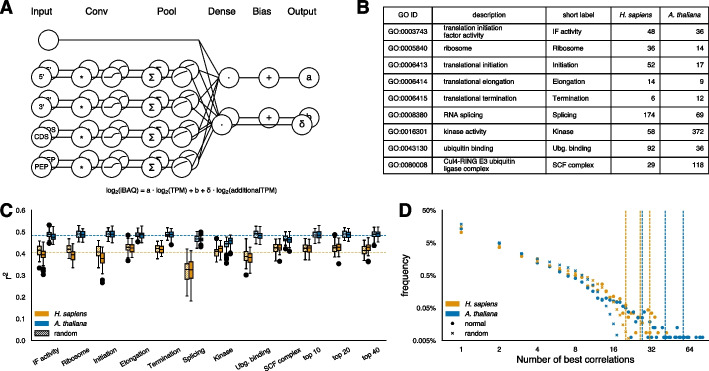
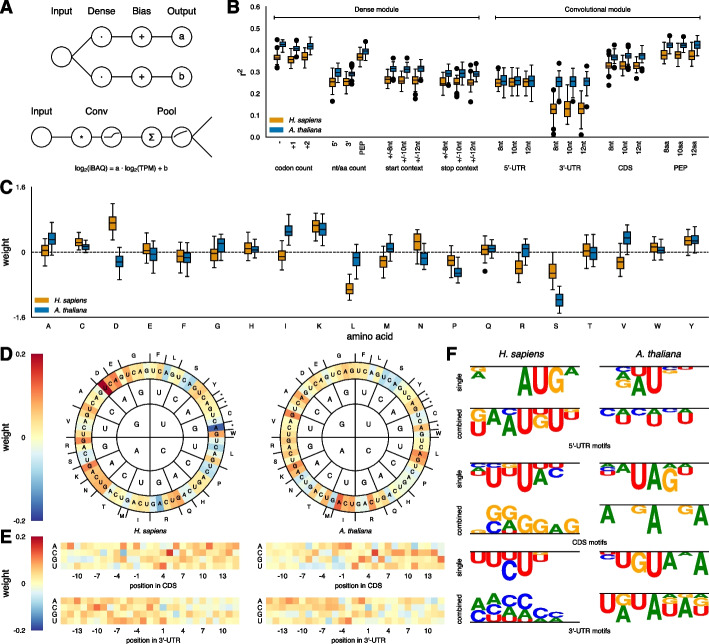
Results: After hyperparameter optimization and initial data exploration, we implemented distinct training modules for value-based and sequence-based data. By analyzing the learned weights, we revealed common and organism-specific sequence features that influence protein-to-mRNA ratios (PTRs), including known and putative sequence motifs. Adding condition-specific protein interaction information identified genes correlated with many PTRs but did not improve predictions, likely due to insufficient data. The integrated model predicted protein abundance on unseen genes with a coefficient of determination (r2) of 0.30 in H. sapiens and 0.32 in A. thaliana.
Conclusions: For H. sapiens, our model improves prediction performance by nearly 50% compared to previous sequence-based approaches, and for A. thaliana it represents the first model of its kind. The model's learned motifs recapitulate known regulatory elements, supporting its utility in systems-level and hypothesis-driven research approaches related to protein regulation.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


