金属-有机骨架结构的数据驱动预测
IF 5.3
2区 化学
Q1 CHEMISTRY, MEDICINAL
Journal of Chemical Information and Modeling
Pub Date : 2025-02-10
DOI:10.1021/acs.jcim.4c0236710.1021/acs.jcim.4c02367
引用次数: 0
摘要
晶体结构预测(CSP)已被证明是发现新材料的有效途径。然而,用于金属有机框架(mof)的CSP的从头算技术不能扩展到高通量模式。在这里,我们提出了一种数据驱动的方法来解决当前计算MOF发现的需求。具体来说,实现了粗粒度神经网络来预测底层网络拓扑。模型得到了令人满意的性能,并通过适用范围的限制进一步增强了模型的性能。本文章由计算机程序翻译,如有差异,请以英文原文为准。
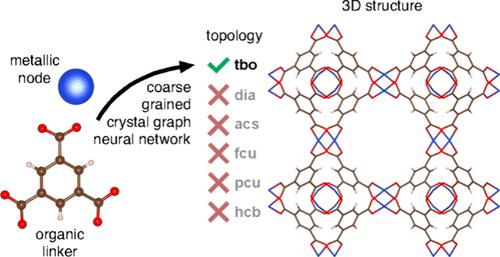
Data-Driven Prediction of Structures of Metal–Organic Frameworks
Crystal structure prediction (CSP) has proven to be an effective route for the discovery of new materials. Nonetheless, the ab initio techniques employed for the CSP of metal–organic frameworks (MOFs) cannot be scaled to a high-throughput mode. Here, we propose a data-driven method for addressing the current needs of computational MOF discovery. Specifically, coarse-grained neural networks were implemented to predict the underlying net topology. The models showed satisfactory performance, which was next enhanced via the limitation of the applicability domain.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
9.80
自引率
10.70%
发文量
529
审稿时长
1.4 months
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


