Huizhi Xie, Han Yu, Shucai Xia*, Bo Wen*, Weiqing Zhang, Zefeng Ren, Xueming Yang and Chuanyao Zhou*,
{"title":"偏振光分辨光电子能谱研究锐钛矿TiO2(101)的各向异性价带结构","authors":"Huizhi Xie, Han Yu, Shucai Xia*, Bo Wen*, Weiqing Zhang, Zefeng Ren, Xueming Yang and Chuanyao Zhou*, ","doi":"10.1021/acs.jpcc.4c0737410.1021/acs.jpcc.4c07374","DOIUrl":null,"url":null,"abstract":"<p >Anatase TiO<sub>2</sub> (A-TiO<sub>2</sub>) has been a prominent material in the field of solar-to-chemical energy conversion due to its outstanding physical and chemical properties, which are closely related to its electronic structure. Nevertheless, the valence band (VB) structure of A-TiO<sub>2</sub>(101), the most stably faceted A-TiO<sub>2</sub>, has seldom been experimentally investigated. In this work, polarization-dependent angle-resolved photoelectron spectroscopy (ARPES) in combination with density functional theory calculations has been used to investigate the VB of A-TiO<sub>2</sub>(101). σ, π, and Pπ bands have been detected in the 4–9 eV binding energy range. The three bands along Γ-Q are rather flat, such that the band center energies vary within a narrow range of 0.5 eV. However, the bands along Γ-M are much more dispersive, displaying significant anisotropy arising from an anisotropic atomic structure. The calculated band structure is in reasonable agreement with the measurements. The π bands along Γ-M exhibit apparent polarization dependence, which is attributed to the different orbital symmetries of the initial states based on the photoemission matrix element effect. The anisotropic valence band structure results in the anisotropic hole effective mass (<i>m</i><sub>h</sub>). The direct measurements of the anisotropic valence band structure of A-TiO<sub>2</sub>(101) will be beneficial to future research and applications of this material.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 7","pages":"3817–3825 3817–3825"},"PeriodicalIF":3.2000,"publicationDate":"2025-02-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Anisotropic Valence Band Structure of Anatase TiO2(101) Studied by Polarization-Dependent Angle-Resolved Photoelectron Spectroscopy\",\"authors\":\"Huizhi Xie, Han Yu, Shucai Xia*, Bo Wen*, Weiqing Zhang, Zefeng Ren, Xueming Yang and Chuanyao Zhou*, \",\"doi\":\"10.1021/acs.jpcc.4c0737410.1021/acs.jpcc.4c07374\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Anatase TiO<sub>2</sub> (A-TiO<sub>2</sub>) has been a prominent material in the field of solar-to-chemical energy conversion due to its outstanding physical and chemical properties, which are closely related to its electronic structure. Nevertheless, the valence band (VB) structure of A-TiO<sub>2</sub>(101), the most stably faceted A-TiO<sub>2</sub>, has seldom been experimentally investigated. In this work, polarization-dependent angle-resolved photoelectron spectroscopy (ARPES) in combination with density functional theory calculations has been used to investigate the VB of A-TiO<sub>2</sub>(101). σ, π, and Pπ bands have been detected in the 4–9 eV binding energy range. The three bands along Γ-Q are rather flat, such that the band center energies vary within a narrow range of 0.5 eV. However, the bands along Γ-M are much more dispersive, displaying significant anisotropy arising from an anisotropic atomic structure. The calculated band structure is in reasonable agreement with the measurements. The π bands along Γ-M exhibit apparent polarization dependence, which is attributed to the different orbital symmetries of the initial states based on the photoemission matrix element effect. The anisotropic valence band structure results in the anisotropic hole effective mass (<i>m</i><sub>h</sub>). The direct measurements of the anisotropic valence band structure of A-TiO<sub>2</sub>(101) will be beneficial to future research and applications of this material.</p>\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"129 7\",\"pages\":\"3817–3825 3817–3825\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-02-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpcc.4c07374\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.4c07374","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Anisotropic Valence Band Structure of Anatase TiO2(101) Studied by Polarization-Dependent Angle-Resolved Photoelectron Spectroscopy
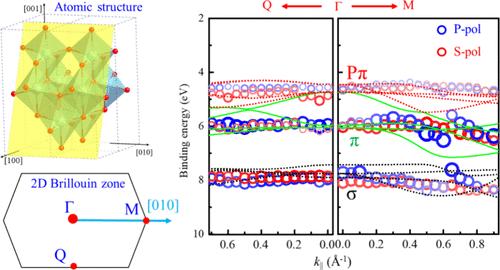
Anatase TiO2 (A-TiO2) has been a prominent material in the field of solar-to-chemical energy conversion due to its outstanding physical and chemical properties, which are closely related to its electronic structure. Nevertheless, the valence band (VB) structure of A-TiO2(101), the most stably faceted A-TiO2, has seldom been experimentally investigated. In this work, polarization-dependent angle-resolved photoelectron spectroscopy (ARPES) in combination with density functional theory calculations has been used to investigate the VB of A-TiO2(101). σ, π, and Pπ bands have been detected in the 4–9 eV binding energy range. The three bands along Γ-Q are rather flat, such that the band center energies vary within a narrow range of 0.5 eV. However, the bands along Γ-M are much more dispersive, displaying significant anisotropy arising from an anisotropic atomic structure. The calculated band structure is in reasonable agreement with the measurements. The π bands along Γ-M exhibit apparent polarization dependence, which is attributed to the different orbital symmetries of the initial states based on the photoemission matrix element effect. The anisotropic valence band structure results in the anisotropic hole effective mass (mh). The direct measurements of the anisotropic valence band structure of A-TiO2(101) will be beneficial to future research and applications of this material.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


