{"title":"MetArea:基于ChIP-seq数据分析转录因子结合位点对基序互斥发生的软件包。","authors":"V G Levitsky, A V Tsukanov, T I Merkulova","doi":"10.18699/vjgb-24-90","DOIUrl":null,"url":null,"abstract":"<p><p>ChIP-seq technology, which is based on chromatin immunoprecipitation (ChIP), allows mapping a set of genomic loci (peaks) containing binding sites (BS) for the investigated (target) transcription factor (TF). A TF may recognize several structurally different BS motifs. The multiprotein complex mapped in a ChIP-seq experiment includes target and other \"partner\" TFs linked by protein-protein interactions. Not all these TFs bind to DNA directly. Therefore, both target and partner TFs recognize enriched BS motifs in peaks. A de novo search approach is used to search for enriched TF BS motifs in ChIP-seq data. For a pair of enriched BS motifs of TFs, the co-occurrence or mutually exclusive occurrence can be detected from a set of peaks: the co-occurrence reflects a more frequent occurrence of two motifs in the same peaks, while the mutually exclusive means their more frequent detection in different peaks. We propose the MetArea software package to identify pairs of TF BS motifs with the mutually exclusive occurrence in ChIP-seq data. MetArea was designed to predict the structural diversity of BS motifs of the same TFs, and the functional relation of BS motifs of different TFs. The functional relation of the motifs of the two distinct TFs presumes that they are interchangeable as part of a multiprotein complex that uses the BS of these TFs to bind directly to DNA in different peaks. MetArea calculates the estimates of recognition performance pAUPRC (partial area under the Precision-Recall curve) for each of the two input single motifs, identifies the \"joint\" motif, and computes the performance for it too. The goal of the analysis is to find pairs of single motifs A and B for which the accuracy of the joint A&B motif is higher than those of both single motifs.</p>","PeriodicalId":44339,"journal":{"name":"Vavilovskii Zhurnal Genetiki i Selektsii","volume":"28 8","pages":"822-833"},"PeriodicalIF":1.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11813801/pdf/","citationCount":"0","resultStr":"{\"title\":\"MetArea: a software package for analysis of the mutually exclusive occurrence in pairs of motifs of transcription factor binding sites based on ChIP-seq data.\",\"authors\":\"V G Levitsky, A V Tsukanov, T I Merkulova\",\"doi\":\"10.18699/vjgb-24-90\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>ChIP-seq technology, which is based on chromatin immunoprecipitation (ChIP), allows mapping a set of genomic loci (peaks) containing binding sites (BS) for the investigated (target) transcription factor (TF). A TF may recognize several structurally different BS motifs. The multiprotein complex mapped in a ChIP-seq experiment includes target and other \\\"partner\\\" TFs linked by protein-protein interactions. Not all these TFs bind to DNA directly. Therefore, both target and partner TFs recognize enriched BS motifs in peaks. A de novo search approach is used to search for enriched TF BS motifs in ChIP-seq data. For a pair of enriched BS motifs of TFs, the co-occurrence or mutually exclusive occurrence can be detected from a set of peaks: the co-occurrence reflects a more frequent occurrence of two motifs in the same peaks, while the mutually exclusive means their more frequent detection in different peaks. We propose the MetArea software package to identify pairs of TF BS motifs with the mutually exclusive occurrence in ChIP-seq data. MetArea was designed to predict the structural diversity of BS motifs of the same TFs, and the functional relation of BS motifs of different TFs. The functional relation of the motifs of the two distinct TFs presumes that they are interchangeable as part of a multiprotein complex that uses the BS of these TFs to bind directly to DNA in different peaks. MetArea calculates the estimates of recognition performance pAUPRC (partial area under the Precision-Recall curve) for each of the two input single motifs, identifies the \\\"joint\\\" motif, and computes the performance for it too. The goal of the analysis is to find pairs of single motifs A and B for which the accuracy of the joint A&B motif is higher than those of both single motifs.</p>\",\"PeriodicalId\":44339,\"journal\":{\"name\":\"Vavilovskii Zhurnal Genetiki i Selektsii\",\"volume\":\"28 8\",\"pages\":\"822-833\"},\"PeriodicalIF\":1.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11813801/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Vavilovskii Zhurnal Genetiki i Selektsii\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.18699/vjgb-24-90\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"AGRICULTURE, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Vavilovskii Zhurnal Genetiki i Selektsii","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.18699/vjgb-24-90","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"AGRICULTURE, MULTIDISCIPLINARY","Score":null,"Total":0}
MetArea: a software package for analysis of the mutually exclusive occurrence in pairs of motifs of transcription factor binding sites based on ChIP-seq data.
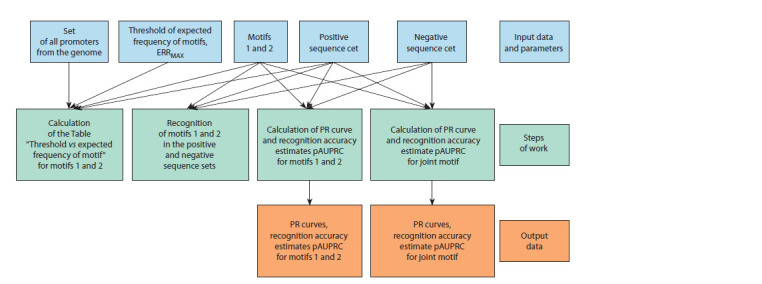
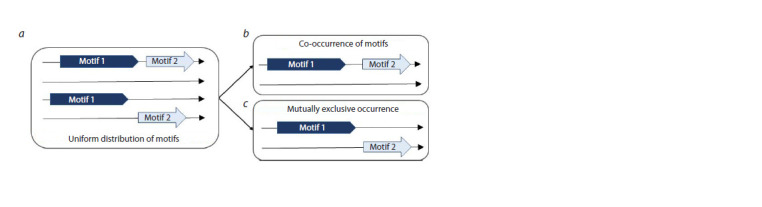
ChIP-seq technology, which is based on chromatin immunoprecipitation (ChIP), allows mapping a set of genomic loci (peaks) containing binding sites (BS) for the investigated (target) transcription factor (TF). A TF may recognize several structurally different BS motifs. The multiprotein complex mapped in a ChIP-seq experiment includes target and other "partner" TFs linked by protein-protein interactions. Not all these TFs bind to DNA directly. Therefore, both target and partner TFs recognize enriched BS motifs in peaks. A de novo search approach is used to search for enriched TF BS motifs in ChIP-seq data. For a pair of enriched BS motifs of TFs, the co-occurrence or mutually exclusive occurrence can be detected from a set of peaks: the co-occurrence reflects a more frequent occurrence of two motifs in the same peaks, while the mutually exclusive means their more frequent detection in different peaks. We propose the MetArea software package to identify pairs of TF BS motifs with the mutually exclusive occurrence in ChIP-seq data. MetArea was designed to predict the structural diversity of BS motifs of the same TFs, and the functional relation of BS motifs of different TFs. The functional relation of the motifs of the two distinct TFs presumes that they are interchangeable as part of a multiprotein complex that uses the BS of these TFs to bind directly to DNA in different peaks. MetArea calculates the estimates of recognition performance pAUPRC (partial area under the Precision-Recall curve) for each of the two input single motifs, identifies the "joint" motif, and computes the performance for it too. The goal of the analysis is to find pairs of single motifs A and B for which the accuracy of the joint A&B motif is higher than those of both single motifs.
期刊介绍:
The "Vavilov Journal of genetics and breeding" publishes original research and review articles in all key areas of modern plant, animal and human genetics, genomics, bioinformatics and biotechnology. One of the main objectives of the journal is integration of theoretical and applied research in the field of genetics. Special attention is paid to the most topical areas in modern genetics dealing with global concerns such as food security and human health.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


