质子化和氧化活化过氧CoIIICoIII配合物的亲电活性:氢过氧、超氧和过氧CoIIICoIII配合物的反应活性和分子结构
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
摘要
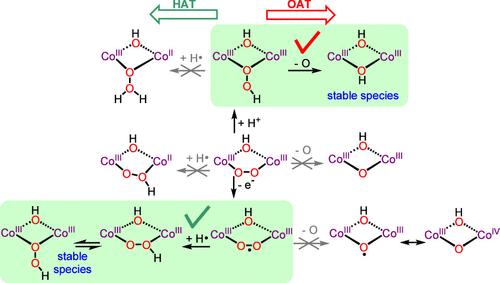
在自然界中,氧化金属酶通常以过氧化物中间体的形式结合O2,这通常需要进一步的质子化激活,因此提出了氢过氧化物物种的形成。在双核金属酶中,通常以铁、锰和铜为特征,过氧、超氧和氢过氧中间体的结构和亲电反应性由于其反应性和瞬态性质通常难以研究,这就是为什么更稳定的过氧CoIIICoIII配合物可能是有用的模型。本文研究了一系列μ-1,2-过氧、μ-1,2-超氧和μ-1,1-氢过氧CoIIICoIII配合物的分子结构及其亲电性氢原子转移(HAT)和氧原子转移(OAT)反应活性的变化。μ-1,2-过氧配合物无亲电活性,而μ-1,2-超氧配合物的氧化激活亲电性HAT反应,而μ-1,1-氢过氧配合物的质子化激活亲电性OAT反应。后者通过μ-1,1-氢过氧配体作为亲电OAT剂的相关机制发生。质子化到μ-1,1-氢过氧使O-O键拉长,而氧化到μ-1,2-超氧使O-O键相对于μ-1,2-过氧缩短。通过在最近报道的μ-1,2-过氧CoIIICoIII配合物中引入远端给电子取代基,μ-1,2-过氧配合物的质子化可以使μ-1,2-过氧的碱度提高5个pKa以上。因此,配体环境最初增加的电子给能增加了μ-1,2-过氧的碱度,使其质子化成更亲电的μ-1,1-氢过氧配体。更一般地说,HAT和OAT反应性与以下中间体的可及性相关。本文章由计算机程序翻译,如有差异,请以英文原文为准。
Activation of Electrophilic Reactivity by Protonation and Oxidation of a Peroxo CoIIICoIII Complex: Reactivity and Molecular Structures of Hydroperoxo, Superoxo, and Peroxo CoIIICoIII Complexes
In nature, oxidizing metalloenzymes usually bind O2 in the form of peroxo intermediates that frequently requires for activation a further protonation, for which the formation of hydroperoxo species is proposed. In dinuclear metalloenzymes, which typically feature Fe, Mn, and Cu, structures and electrophilic reactivity of peroxo, superoxo, and hydroperoxo intermediates are generally difficult to study due to their reactive and transient nature, which is why more stable peroxo CoIIICoIII complexes might be helpful models. Here, we present the molecular structures of a series of μ-1,2-peroxo, μ-1,2-superoxo, and μ-1,1-hydroperoxo CoIIICoIII complexes and the variation of their electrophilic hydrogen-atom-transfer (HAT) and oxygen-atom-transfer (OAT) reactivity. The μ-1,2-peroxo complex exhibits no electrophilic reactivity, whereas oxidation to the μ-1,2-superoxo complex activates electrophilic HAT reactivity, while protonation to the μ-1,1-hydroperoxo complex activates electrophilic OAT reactivity. The latter occurs by an associated mechanism with the μ-1,1-hydroperoxo ligand being the electrophilic OAT agent. Protonation to the μ-1,1-hydroperoxo elongates the O–O bond, while oxidation to the μ-1,2-superoxo shortens it relative to the μ-1,2-peroxo. The protonation of the μ-1,2-peroxo complex could be made accessible by the introduction of remote electron-donating substituents into a recently reported μ-1,2-peroxo CoIIICoIII complex that increases the μ-1,2-peroxo basicity by more than five pKa units. Hence, the initially counterintuitive increase of electron donation by the ligand environment increases the μ-1,2-peroxo basicity allowing its protonation to a more electrophilic μ-1,1-hydroperoxo ligand. More generally, HAT and OAT reactivity is correlated with accessibility of the following intermediate.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


