Timothé Rouzé, Igor Martayan, Camille Marchet, Antoine Limasset
{"title":"分数命中集用于高效的多集素描。","authors":"Timothé Rouzé, Igor Martayan, Camille Marchet, Antoine Limasset","doi":"10.1186/s13015-024-00268-0","DOIUrl":null,"url":null,"abstract":"<p><p>The exponential increase in publicly available sequencing data and genomic resources necessitates the development of highly efficient methods for data processing and analysis. Locality-sensitive hashing techniques have successfully transformed large datasets into smaller, more manageable sketches while maintaining comparability using metrics such as Jaccard and containment indices. However, fixed-size sketches encounter difficulties when applied to divergent datasets. Scalable sketching methods, such as sourmash, provide valuable solutions but still lack resource-efficient, tailored indexing. Our objective is to create lighter sketches with comparable results while enhancing efficiency. We introduce the concept of Fractional Hitting Sets, a generalization of Universal Hitting Sets, which cover a specified fraction of the k-mer space. In theory and practice, we demonstrate the feasibility of achieving such coverage with simple but highly efficient schemes. By encoding the covered k-mers as super-k-mers, we provide a space-efficient exact representation that also enables optimized comparisons. Our novel tool, supersampler, implements this scheme, and experimental results with real bacterial collections closely match our theoretical findings. In comparison to sourmash, supersampler achieves similar outcomes while utilizing an order of magnitude less space and memory and operating several times faster. This highlights the potential of our approach in addressing the challenges presented by the ever-expanding landscape of genomic data. supersampler is an open-source software and can be accessed at https://github.com/TimRouze/supersampler . The data required to reproduce the results presented in this manuscript is available at https://github.com/TimRouze/supersampler/experiments .</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"20 1","pages":"1"},"PeriodicalIF":1.7000,"publicationDate":"2025-02-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11807336/pdf/","citationCount":"0","resultStr":"{\"title\":\"Fractional hitting sets for efficient multiset sketching.\",\"authors\":\"Timothé Rouzé, Igor Martayan, Camille Marchet, Antoine Limasset\",\"doi\":\"10.1186/s13015-024-00268-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>The exponential increase in publicly available sequencing data and genomic resources necessitates the development of highly efficient methods for data processing and analysis. Locality-sensitive hashing techniques have successfully transformed large datasets into smaller, more manageable sketches while maintaining comparability using metrics such as Jaccard and containment indices. However, fixed-size sketches encounter difficulties when applied to divergent datasets. Scalable sketching methods, such as sourmash, provide valuable solutions but still lack resource-efficient, tailored indexing. Our objective is to create lighter sketches with comparable results while enhancing efficiency. We introduce the concept of Fractional Hitting Sets, a generalization of Universal Hitting Sets, which cover a specified fraction of the k-mer space. In theory and practice, we demonstrate the feasibility of achieving such coverage with simple but highly efficient schemes. By encoding the covered k-mers as super-k-mers, we provide a space-efficient exact representation that also enables optimized comparisons. Our novel tool, supersampler, implements this scheme, and experimental results with real bacterial collections closely match our theoretical findings. In comparison to sourmash, supersampler achieves similar outcomes while utilizing an order of magnitude less space and memory and operating several times faster. This highlights the potential of our approach in addressing the challenges presented by the ever-expanding landscape of genomic data. supersampler is an open-source software and can be accessed at https://github.com/TimRouze/supersampler . The data required to reproduce the results presented in this manuscript is available at https://github.com/TimRouze/supersampler/experiments .</p>\",\"PeriodicalId\":50823,\"journal\":{\"name\":\"Algorithms for Molecular Biology\",\"volume\":\"20 1\",\"pages\":\"1\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-02-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11807336/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Algorithms for Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13015-024-00268-0\",\"RegionNum\":4,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-024-00268-0","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Fractional hitting sets for efficient multiset sketching.
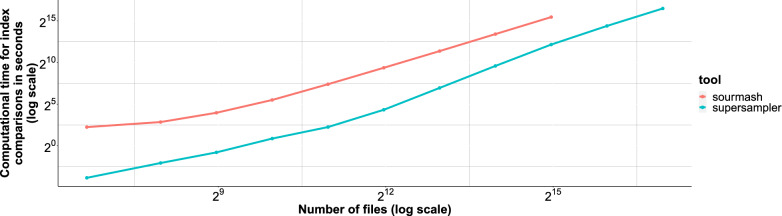
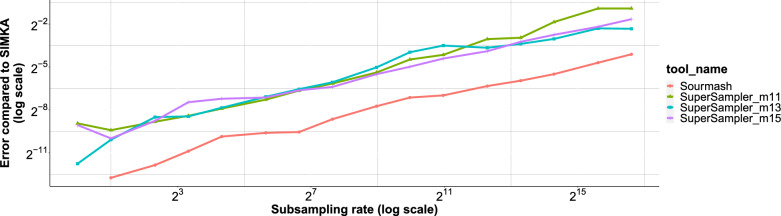
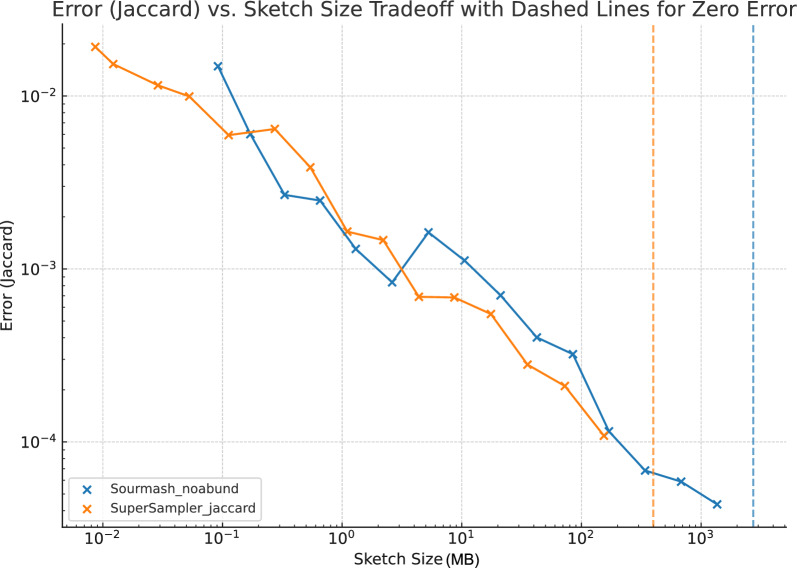
The exponential increase in publicly available sequencing data and genomic resources necessitates the development of highly efficient methods for data processing and analysis. Locality-sensitive hashing techniques have successfully transformed large datasets into smaller, more manageable sketches while maintaining comparability using metrics such as Jaccard and containment indices. However, fixed-size sketches encounter difficulties when applied to divergent datasets. Scalable sketching methods, such as sourmash, provide valuable solutions but still lack resource-efficient, tailored indexing. Our objective is to create lighter sketches with comparable results while enhancing efficiency. We introduce the concept of Fractional Hitting Sets, a generalization of Universal Hitting Sets, which cover a specified fraction of the k-mer space. In theory and practice, we demonstrate the feasibility of achieving such coverage with simple but highly efficient schemes. By encoding the covered k-mers as super-k-mers, we provide a space-efficient exact representation that also enables optimized comparisons. Our novel tool, supersampler, implements this scheme, and experimental results with real bacterial collections closely match our theoretical findings. In comparison to sourmash, supersampler achieves similar outcomes while utilizing an order of magnitude less space and memory and operating several times faster. This highlights the potential of our approach in addressing the challenges presented by the ever-expanding landscape of genomic data. supersampler is an open-source software and can be accessed at https://github.com/TimRouze/supersampler . The data required to reproduce the results presented in this manuscript is available at https://github.com/TimRouze/supersampler/experiments .
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


