Gaopeng Shi*, Jianjun Zhang, Xu Wang and Yangyang Xu*,
{"title":"分子间相互作用和链刚度对丙烯酸网络玻璃化形成的复杂影响","authors":"Gaopeng Shi*, Jianjun Zhang, Xu Wang and Yangyang Xu*, ","doi":"10.1021/acs.macromol.4c0271210.1021/acs.macromol.4c02712","DOIUrl":null,"url":null,"abstract":"<p >The knowledge of molecular dynamics induced by noncovalent bonding is important for the fabrication of high-performance polymeric materials. However, little attention is paid to the segmental dynamics triggered by the π–π stacking interactions and fluorinated polar interactions. Here, the complicated effects of rigidity and intermolecular interaction on the dynamic fragility <i>m</i> and glass transition temperature <i>T</i><sub>g</sub> are systematically investigated in acrylic networks. It is found that fluorinated structures have minor effects on <i>m</i> and <i>T</i><sub>g</sub> due to their relatively weak intermolecular interaction strength, whereas <i>m</i> increases with the growth of <i>T</i><sub>g</sub> after the introduction of rigid aromatic structures. In contrast, the further addition of hindered phenols or metallic ions leads to an increased <i>T</i><sub>g</sub> while <i>m</i> is reduced. Based on the generalized entropy theory (GET) of glass formation, it can be concluded that polymers with rigid structures but weak interaction strength tend to be fragile due to the almost constant cohesive energy densities. Although the hydrogen bonding and ionic interactions act as transient cross-linkers, the cohesive energy densities also increase because of their high polarities, thus leading to decreased fragilities. These results not only help further clarify the relationship between structures and dynamics on glass formation but also provide fundamental experimental benefits for verifying the predictions of GET and molecular dynamic simulations of glass-forming polymer materials.</p>","PeriodicalId":51,"journal":{"name":"Macromolecules","volume":"58 1","pages":"187–198 187–198"},"PeriodicalIF":5.2000,"publicationDate":"2025-01-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Complicated Effects of Intermolecular Interaction and Chain Rigidity on the Glass Formation of Acrylic Networks\",\"authors\":\"Gaopeng Shi*, Jianjun Zhang, Xu Wang and Yangyang Xu*, \",\"doi\":\"10.1021/acs.macromol.4c0271210.1021/acs.macromol.4c02712\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The knowledge of molecular dynamics induced by noncovalent bonding is important for the fabrication of high-performance polymeric materials. However, little attention is paid to the segmental dynamics triggered by the π–π stacking interactions and fluorinated polar interactions. Here, the complicated effects of rigidity and intermolecular interaction on the dynamic fragility <i>m</i> and glass transition temperature <i>T</i><sub>g</sub> are systematically investigated in acrylic networks. It is found that fluorinated structures have minor effects on <i>m</i> and <i>T</i><sub>g</sub> due to their relatively weak intermolecular interaction strength, whereas <i>m</i> increases with the growth of <i>T</i><sub>g</sub> after the introduction of rigid aromatic structures. In contrast, the further addition of hindered phenols or metallic ions leads to an increased <i>T</i><sub>g</sub> while <i>m</i> is reduced. Based on the generalized entropy theory (GET) of glass formation, it can be concluded that polymers with rigid structures but weak interaction strength tend to be fragile due to the almost constant cohesive energy densities. Although the hydrogen bonding and ionic interactions act as transient cross-linkers, the cohesive energy densities also increase because of their high polarities, thus leading to decreased fragilities. These results not only help further clarify the relationship between structures and dynamics on glass formation but also provide fundamental experimental benefits for verifying the predictions of GET and molecular dynamic simulations of glass-forming polymer materials.</p>\",\"PeriodicalId\":51,\"journal\":{\"name\":\"Macromolecules\",\"volume\":\"58 1\",\"pages\":\"187–198 187–198\"},\"PeriodicalIF\":5.2000,\"publicationDate\":\"2025-01-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Macromolecules\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.macromol.4c02712\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"POLYMER SCIENCE\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Macromolecules","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.macromol.4c02712","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"POLYMER SCIENCE","Score":null,"Total":0}
Complicated Effects of Intermolecular Interaction and Chain Rigidity on the Glass Formation of Acrylic Networks
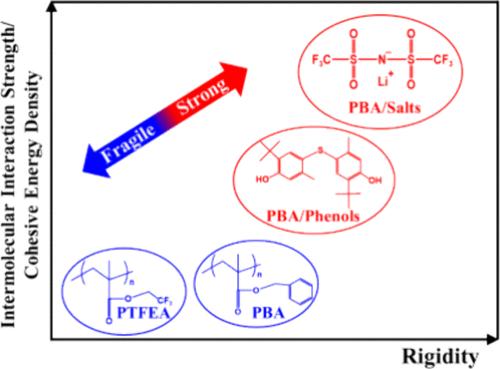
The knowledge of molecular dynamics induced by noncovalent bonding is important for the fabrication of high-performance polymeric materials. However, little attention is paid to the segmental dynamics triggered by the π–π stacking interactions and fluorinated polar interactions. Here, the complicated effects of rigidity and intermolecular interaction on the dynamic fragility m and glass transition temperature Tg are systematically investigated in acrylic networks. It is found that fluorinated structures have minor effects on m and Tg due to their relatively weak intermolecular interaction strength, whereas m increases with the growth of Tg after the introduction of rigid aromatic structures. In contrast, the further addition of hindered phenols or metallic ions leads to an increased Tg while m is reduced. Based on the generalized entropy theory (GET) of glass formation, it can be concluded that polymers with rigid structures but weak interaction strength tend to be fragile due to the almost constant cohesive energy densities. Although the hydrogen bonding and ionic interactions act as transient cross-linkers, the cohesive energy densities also increase because of their high polarities, thus leading to decreased fragilities. These results not only help further clarify the relationship between structures and dynamics on glass formation but also provide fundamental experimental benefits for verifying the predictions of GET and molecular dynamic simulations of glass-forming polymer materials.
期刊介绍:
Macromolecules publishes original, fundamental, and impactful research on all aspects of polymer science. Topics of interest include synthesis (e.g., controlled polymerizations, polymerization catalysis, post polymerization modification, new monomer structures and polymer architectures, and polymerization mechanisms/kinetics analysis); phase behavior, thermodynamics, dynamic, and ordering/disordering phenomena (e.g., self-assembly, gelation, crystallization, solution/melt/solid-state characteristics); structure and properties (e.g., mechanical and rheological properties, surface/interfacial characteristics, electronic and transport properties); new state of the art characterization (e.g., spectroscopy, scattering, microscopy, rheology), simulation (e.g., Monte Carlo, molecular dynamics, multi-scale/coarse-grained modeling), and theoretical methods. Renewable/sustainable polymers, polymer networks, responsive polymers, electro-, magneto- and opto-active macromolecules, inorganic polymers, charge-transporting polymers (ion-containing, semiconducting, and conducting), nanostructured polymers, and polymer composites are also of interest. Typical papers published in Macromolecules showcase important and innovative concepts, experimental methods/observations, and theoretical/computational approaches that demonstrate a fundamental advance in the understanding of polymers.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


