Asim Sajjad, Muhammad Faizan, Tahani A. Alrebdi, Ghulam Murtaza, Javed Rehman, Xingchen Shen, Yujing Dong, Kausar Shaheen and Shah Haidar Khan
{"title":"双钙钛矿Cs2AgSbX6 (X = Cl, Br, I)作为光电子和热电材料的第一性原理研究","authors":"Asim Sajjad, Muhammad Faizan, Tahani A. Alrebdi, Ghulam Murtaza, Javed Rehman, Xingchen Shen, Yujing Dong, Kausar Shaheen and Shah Haidar Khan","doi":"10.1039/D4CP04662E","DOIUrl":null,"url":null,"abstract":"<p >Perovskite compounds have been well explored, and they are suitable for solar energy as well as thermoelectric applications. Herein, we present the structural, electronic, optical, thermoelectric, and elastic properties of Cs<small><sub>2</sub></small>AgSbX<small><sub>6</sub></small> (X = Cl, Br, and I) perovskites with the help of density functional theory (DFT). The Wu-Cohen generalized gradient approximation (WC-GGA) and Perdew–Burke–Ernzerhof GGA (PBE-GGA) were used to calculate the structural parameters. According to the cohesive energy, phonon spectrum, and AIMD simulation results, Cs<small><sub>2</sub></small>AgSbX<small><sub>6</sub></small> demonstrated good thermodynamic, dynamical, and thermal stabilities. The compounds exhibited an indirect band gap according to the band structure results obtained <em>via</em> the PBE and Heyd–Scuseria–Ernzerhof (HSE06) functionals. The band gap were 2.28, 1.63, and 0.99 eV for <strong>Cs<small><sub>2</sub></small>AgSbCl<small><sub>6</sub></small></strong>, <strong>Cs<small><sub>2</sub></small>AgSbBr<small><sub>6</sub></small></strong>, and <strong>Cs<small><sub>2</sub></small>AgSbI<small><sub>6</sub></small></strong>, respectively. Optical absorption (6.05 × 10<small><sup>5</sup></small> cm<small><sup>−1</sup></small>) and spectroscopic limited maximum efficiency (SLME > 30%) calculations revealed that <strong>Cs<small><sub>2</sub></small>AgSbI<small><sub>6</sub></small></strong> worked effectively in the visible range. The figure of merit (<em>ZT</em>) calculations showed an increasing trend with increasing temperature. The maximum values were ∼0.77 (500 K), 0.76 (700 K), and 0.76 (750 K) for <strong>Cs<small><sub>2</sub></small>AgSbCl<small><sub>6</sub></small></strong>, <strong>Cs<small><sub>2</sub></small>AgSbBr<small><sub>6</sub></small></strong> and <strong>Cs<small><sub>2</sub></small>AgSbI<small><sub>6</sub></small></strong>, respectively. The elastic stability for the selected compounds was confirmed <em>via</em> “Born stability criteria” and found well consistent with the values for cubic materials. Further, Cauchy's pressure (<em>C</em><small><sub>12</sub></small> − <em>C</em><small><sub>44</sub></small>) and Pugh's ratio (<em>B</em>/<em>G</em>) indicated the ductile nature of these compounds. The appropriate band gap, optimum <em>ZT</em>, and thermal and mechanical stabilities suggest the suitability of these compounds for use in optoelectronic and thermoelectric applications.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 9","pages":" 4880-4891"},"PeriodicalIF":2.9000,"publicationDate":"2025-02-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring double perovskites Cs2AgSbX6 (X = Cl, Br, and I) as promising optoelectronic and thermoelectric materials: a first-principles study†\",\"authors\":\"Asim Sajjad, Muhammad Faizan, Tahani A. Alrebdi, Ghulam Murtaza, Javed Rehman, Xingchen Shen, Yujing Dong, Kausar Shaheen and Shah Haidar Khan\",\"doi\":\"10.1039/D4CP04662E\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Perovskite compounds have been well explored, and they are suitable for solar energy as well as thermoelectric applications. Herein, we present the structural, electronic, optical, thermoelectric, and elastic properties of Cs<small><sub>2</sub></small>AgSbX<small><sub>6</sub></small> (X = Cl, Br, and I) perovskites with the help of density functional theory (DFT). The Wu-Cohen generalized gradient approximation (WC-GGA) and Perdew–Burke–Ernzerhof GGA (PBE-GGA) were used to calculate the structural parameters. According to the cohesive energy, phonon spectrum, and AIMD simulation results, Cs<small><sub>2</sub></small>AgSbX<small><sub>6</sub></small> demonstrated good thermodynamic, dynamical, and thermal stabilities. The compounds exhibited an indirect band gap according to the band structure results obtained <em>via</em> the PBE and Heyd–Scuseria–Ernzerhof (HSE06) functionals. The band gap were 2.28, 1.63, and 0.99 eV for <strong>Cs<small><sub>2</sub></small>AgSbCl<small><sub>6</sub></small></strong>, <strong>Cs<small><sub>2</sub></small>AgSbBr<small><sub>6</sub></small></strong>, and <strong>Cs<small><sub>2</sub></small>AgSbI<small><sub>6</sub></small></strong>, respectively. Optical absorption (6.05 × 10<small><sup>5</sup></small> cm<small><sup>−1</sup></small>) and spectroscopic limited maximum efficiency (SLME > 30%) calculations revealed that <strong>Cs<small><sub>2</sub></small>AgSbI<small><sub>6</sub></small></strong> worked effectively in the visible range. The figure of merit (<em>ZT</em>) calculations showed an increasing trend with increasing temperature. The maximum values were ∼0.77 (500 K), 0.76 (700 K), and 0.76 (750 K) for <strong>Cs<small><sub>2</sub></small>AgSbCl<small><sub>6</sub></small></strong>, <strong>Cs<small><sub>2</sub></small>AgSbBr<small><sub>6</sub></small></strong> and <strong>Cs<small><sub>2</sub></small>AgSbI<small><sub>6</sub></small></strong>, respectively. The elastic stability for the selected compounds was confirmed <em>via</em> “Born stability criteria” and found well consistent with the values for cubic materials. Further, Cauchy's pressure (<em>C</em><small><sub>12</sub></small> − <em>C</em><small><sub>44</sub></small>) and Pugh's ratio (<em>B</em>/<em>G</em>) indicated the ductile nature of these compounds. The appropriate band gap, optimum <em>ZT</em>, and thermal and mechanical stabilities suggest the suitability of these compounds for use in optoelectronic and thermoelectric applications.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 9\",\"pages\":\" 4880-4891\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2025-02-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04662e\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04662e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Exploring double perovskites Cs2AgSbX6 (X = Cl, Br, and I) as promising optoelectronic and thermoelectric materials: a first-principles study†
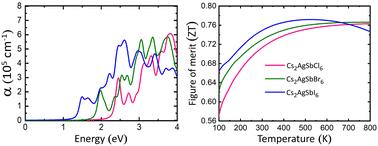
Perovskite compounds have been well explored, and they are suitable for solar energy as well as thermoelectric applications. Herein, we present the structural, electronic, optical, thermoelectric, and elastic properties of Cs2AgSbX6 (X = Cl, Br, and I) perovskites with the help of density functional theory (DFT). The Wu-Cohen generalized gradient approximation (WC-GGA) and Perdew–Burke–Ernzerhof GGA (PBE-GGA) were used to calculate the structural parameters. According to the cohesive energy, phonon spectrum, and AIMD simulation results, Cs2AgSbX6 demonstrated good thermodynamic, dynamical, and thermal stabilities. The compounds exhibited an indirect band gap according to the band structure results obtained via the PBE and Heyd–Scuseria–Ernzerhof (HSE06) functionals. The band gap were 2.28, 1.63, and 0.99 eV for Cs2AgSbCl6, Cs2AgSbBr6, and Cs2AgSbI6, respectively. Optical absorption (6.05 × 105 cm−1) and spectroscopic limited maximum efficiency (SLME > 30%) calculations revealed that Cs2AgSbI6 worked effectively in the visible range. The figure of merit (ZT) calculations showed an increasing trend with increasing temperature. The maximum values were ∼0.77 (500 K), 0.76 (700 K), and 0.76 (750 K) for Cs2AgSbCl6, Cs2AgSbBr6 and Cs2AgSbI6, respectively. The elastic stability for the selected compounds was confirmed via “Born stability criteria” and found well consistent with the values for cubic materials. Further, Cauchy's pressure (C12 − C44) and Pugh's ratio (B/G) indicated the ductile nature of these compounds. The appropriate band gap, optimum ZT, and thermal and mechanical stabilities suggest the suitability of these compounds for use in optoelectronic and thermoelectric applications.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


